San Diego, CA, USA, 11email: vbafna@eng.ucsd.edu, 22institutetext: Bioinformatics Undergraduate Program, Department of Bioengineering,
University of California, San Diego, CA, USA
Cerulean: A hybrid assembly using high throughput short and long reads
Abstract
Genome assembly using high throughput data with short reads, arguably, remains an unresolvable task in repetitive genomes, since when the length of a repeat exceeds the read length, it becomes difficult to unambiguously connect the flanking regions. The emergence of third generation sequencing (Pacific Biosciences) with long reads enables the opportunity to resolve complicated repeats that could not be resolved by the short read data. However, these long reads have high error rate and it is an uphill task to assemble the genome without using additional high quality short reads. Recently, Koren et al. 2012 [1] proposed an approach to use high quality short reads data to correct these long reads and, thus, make the assembly from long reads possible. However, due to the large size of both dataset (short and long reads), error-correction of these long reads requires excessively high computational resources, even on small bacterial genomes. In this work, instead of error correction of long reads, we first assemble the short reads and later map these long reads on the assembly graph to resolve repeats.
Contribution: We present a hybrid assembly approach that is both computationally effective and produces high quality assemblies. Our algorithm first operates with a simplified version of the assembly graph consisting only of long contigs and gradually improves the assembly by adding smaller contigs in each iteration. In contrast to the state-of-the-art long reads error correction technique, which requires high computational resources and long running time on a supercomputer even for bacterial genome datasets, our software can produce comparable assembly using only a standard desktop in a short running time.
1 Introduction
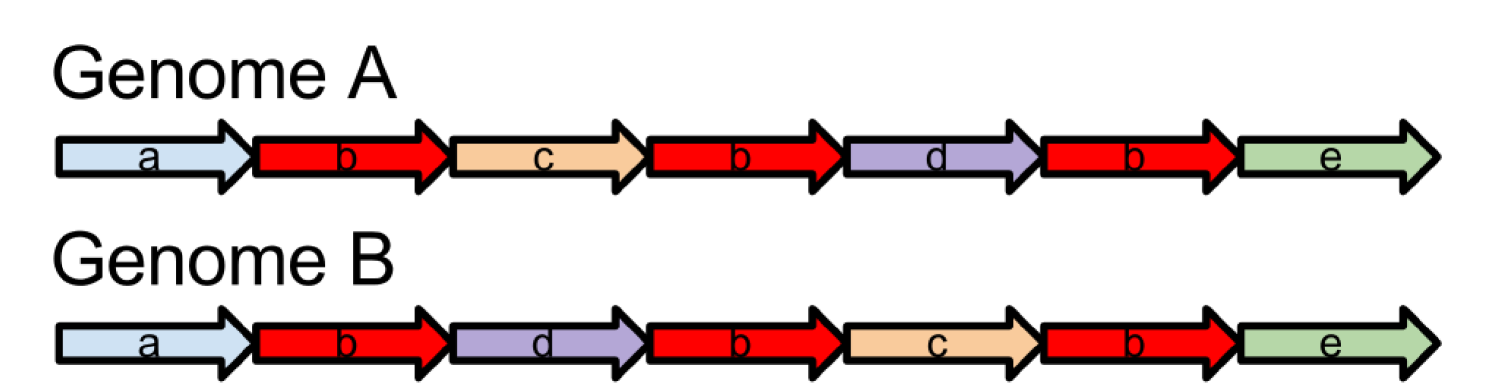
The advent of high throughput sequencing technologies has generated a lot of interest from the computational perspective of de novo assembly of genomic sequences. A major breakthrough in the massively parallel high-throughput sequencing technologies includes the second generation sequencing platforms including those from Illumina and Life Technologies. These platforms generate paired-end reads with length of the order of 100 or 250 base pairs. The mean end-to-end distance between the paired-end reads, called the insert size, is around 300 to 500 base-pairs. The paired-end reads can be sequenced with high accuracy and high depth of coverage. Two approaches are broadly used by assembly tools to assemble the paired-end reads into complete genomes of genomic contigs: (i) Overlap-layout-consensus assembly was introduced by Staden [2], which was later improvised by the introduction of string graph by Myers [3] (Celera Assembler [4], SGA [5]); (ii) de Bruijn graph-based assembly was originally proposed by Idury and Waterman [6] and extended by Pevzner et al [7] (Euler-SR [8], ABySS [9], Velvet [10]). These de novo assembly approaches can generate high quality assemblies using the high quality paired-end reads. However, these paired-end reads are unable to span large repeats and as a result the assembled contigs are often short. Figure 1 shows an example where the assembler is unable to identify the true genome architecture using only short read.
Newer high throughput sequencing platforms [11] target the length limitation of second generation short reads by generating libraries with a large span. The prominent technologies include: (i) jumping libraries which generate small mate-pair reads of around 150 base pairs and variable span of 5 kbp or more; (ii) long reads from Pacific Biosciences with variable length ranging from 1 kbp to 20 kbp and (iii) genomic fragments amplified by Moleculo technology (size: 1.5 kbp to 15 kbp [12]) and then sequenced using short read sequencing technologies. In this work we focus on PacBio long reads generated by directly sequencing entire genomic fragments. In the rest of this article we will use the term short reads to describe the paired-end reads from Illumina and long reads to describe reads generated using the Pacific Biosciences RS platform.
Long reads generated from PacBio RS can easily span most repeats and have the potential to produce very large assembled contigs. Unfortunately, these reads can have a very high error rate with mean error rate as high as 16%. Hence they are difficult to assemble by themselves and require very high coverage; the assembly quality falls rapidly with smaller coverage [13]. However, combining the high quality of second generation short reads and the large length of the long reads, these datasets can be processed simultaneously to produce very long genomic contigs that otherwise required costly low-throughput techniques.
Recent efforts (PacbioToCA [1], LSC [14]) have focused on mapping short reads to the erroneous long reads to correct the long reads using aligners like NovoAlign [15] and GMAP [16] which can allow large edit distance for the mapping. These corrected long reads are then used to generate an assembly with significantly longer contigs. However, such mapping from all short reads to all long reads with large edit-distance is computationally expensive and requires a large running time even for small bacterial datasets. Furthermore, if there are two or more similar regions in the genome, the short reads from one region can still map to long reads from the other region given the high edit-distance. Reads corrected in such fashion may create spurious adjacencies leading to misassemblies.
An alternative approach is to first assemble the high coverage short read dataset to produce high quality contigs and use long reads for scaffolding. Previous tools that use this approach include AHA scaffolder [17] and ALLPATHS-LG [18]. However, these approaches are specialized to perform hybrid assembly in the presence additional libraries including Roche 454 reads for AHA scaffolder and jumping libraries of mate-pair reads for ALLPATHS-LG. Following this approach, a hybrid assembler will essentially take as input: the assembler should find the correct traversal of genome on the graph using the support information from the mapping of long reads.
While the alignments of long reads on the complete genome can be easily identified using BLASR [19], alignments to shorter contigs can be spurious. This is because we have to allow very short alignments and cannot conclusively say if these are true alignments or accidental alignments due to short repeats and similar-looking regions. Such alignments generate ambiguous adjacencies between contigs and stop us from making high confidence calls while scaffolding. Furthermore, as we see in Figure 2, the longer assembled contigs tend to be unique in the genome whereas the shorter contigs tend to repeat. If such short contigs occur adjacent to each other in the genome, then using long reads to determine the exact layout of all the contigs in the presence of spurious alignments becomes a difficult problem.
Contribution: In this work, we present Cerulean, a completely automated hybrid assembly approach to produce a high quality scaffolds using Illumina paired-end reads and PacBio long reads. Cerulean does not use the short reads directly; instead it works with an assembly graph structure generated from short read data using existing assemblers. Assembly graphs are graphs where nodes correspond to contigs of assembled short reads and edges represent putative adjacencies of the contigs, but not confined to overlapping contigs. Such assembly graph are commonly built using overlap-layout-consensus and more recently with the de Bruijn graph paradigm. The input to Cerulean includes: (i) the assembly graph generated by ABySS paired-end read assembler [9] constructed from short reads (and it can be applied to graphs generated by overlap graph or de Bruijn graph based assemblers as long as the graph has the desired format) and (ii) the mapping of long reads to the assembled contigs. The output of Cerulean is an simplified representation of the unentangled assembly graph. The non-branching paths in this simplified graph correspond to the scaffolds in the genome.
We recognize that multiple spurious alignments of long reads to short contigs makes it a difficult problem to unentangle the assembly graph, especially when the short contigs form densely connected substructures. The Cerulean algorithm addresses this problem through an iterative framework to identify and extend high confidence genomic paths. Cerulean initially operates with a simplified representation of the assembly graph, which we call the skeleton graph (Figure 3(a)), consisting only of long contigs. We then gradually improve the assembly by adding smaller contigs to the skeleton graph in each iteration.
Our software produced higher quality assembly than the state-of-the-art software for hybrid assembly (PacbioToCA, AHA) in much shorter running time and lower memory usage. While assembly of Cerulean was significantly better than AHA scaffolder; PacbioToCA required 8 hours to run on a supercomputer with 24 threads for a bacterial genome dataset with a total memory usage of 55GB and temporary files of 300GB. In contrast, Cerulean finished within few minutes on a single thread on a regular desktop with memory usage of 100MB and preprocessing (ABySS, BLASR) taking less than an hour on a desktop computer. Starting with N75 of 60 kbp of contigs generated by ABySS, scaffolds generates scaffolds with N75 of 503 kbp as compared to 247 kbp for PacbioToCA and 106 kbp for AHA scaffolder.
2 Methods
Inputs: The inputs to Cerulean include (i) the assembly graph and contig sequences from short read assembly (using ABySS or other assemblers) and (ii) alignments of long reads to contigs from the assembly graph (using BLASR). The assembly graph consists of one vertex for each contig and a conjugate vertex for its reverse complement. The length of a vertex corresponds to the length of the contig. A directed edge between two vertices indicates a putative adjacency between the two vertices. For every directed edge, the conjugate edge is a directed edge from the conjugate of its sink to the conjugate of its source. For each edge, we define the length to be the offset between the end of source contig and start of the sink contig. Thus, if the two contigs overlap, then the length is a negative number; in case of a gap this length is a positive number. The size of the overlap or the gap may depend on the short read assembler, e.g., if the assembler just produces the de Bruijn graph, then the overlap is directly determined by -mer size, but many short read assemblers also implement preliminary analysis of the graph structure and so the assembled contigs may even overlap by a few thousand base pairs even though the paired-end insert size is only few hundred based pairs. Henceforth, contigs contigs refer to DNA sequences assembled by the short read assembler and scaffolds refer chain of contigs glued together using alignments of long reads to contigs.
Pipeline: The contigs generated by the short paired-end read assembly have a large distribution of lengths and some of these contigs repeat multiple times in the reference. Most repeats tend to be short and there are very few long contigs which occur multiple times in the reference as shown in Figure 2 for E. Coli dataset. Resolution of the assembly graph in the presence of these short and repetitive contigs is difficult since they create noise in mapping (spurious alignments) and may form dense structures in the graph which is a major obstacle for the repeat resolution procedure.
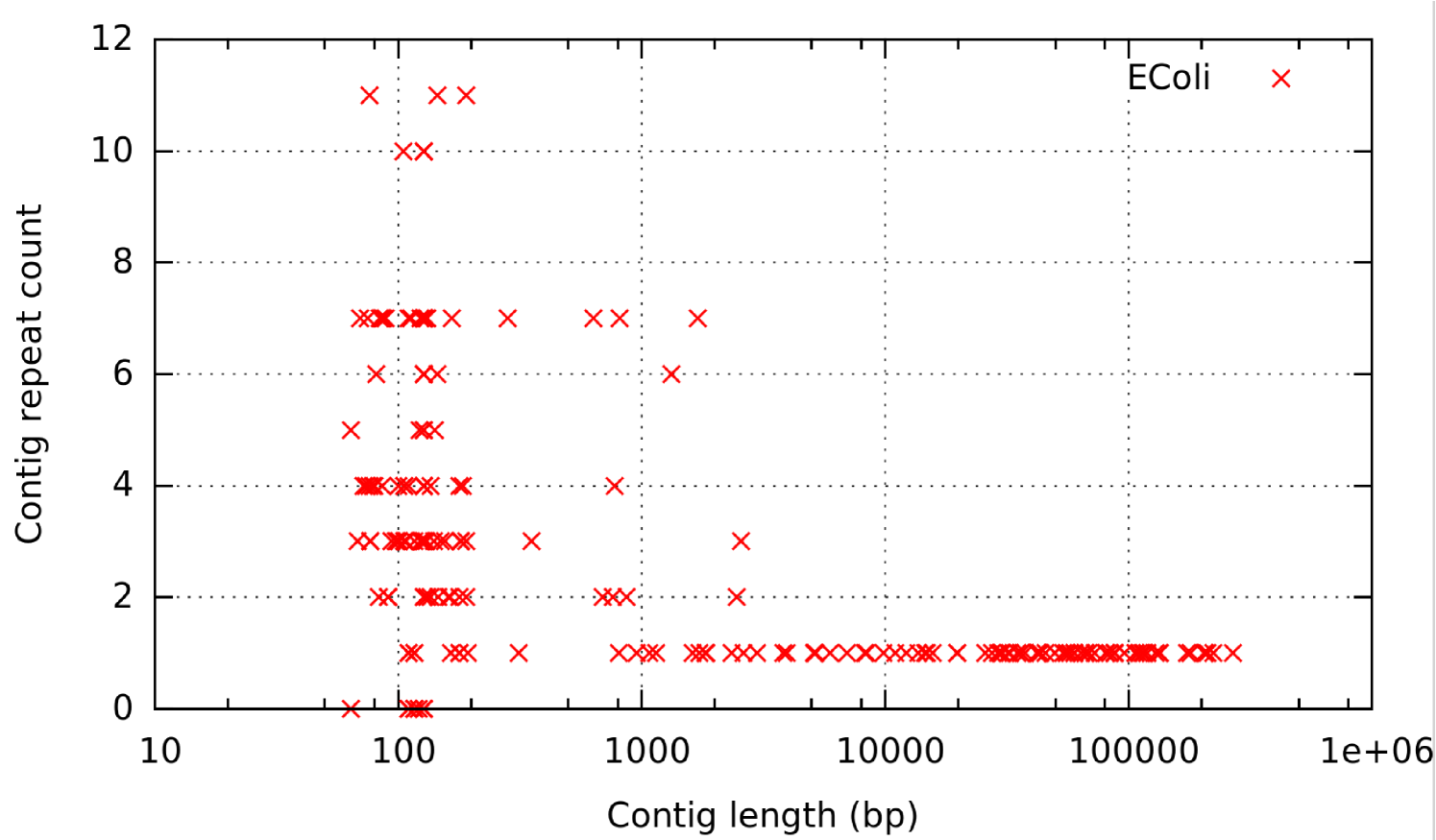
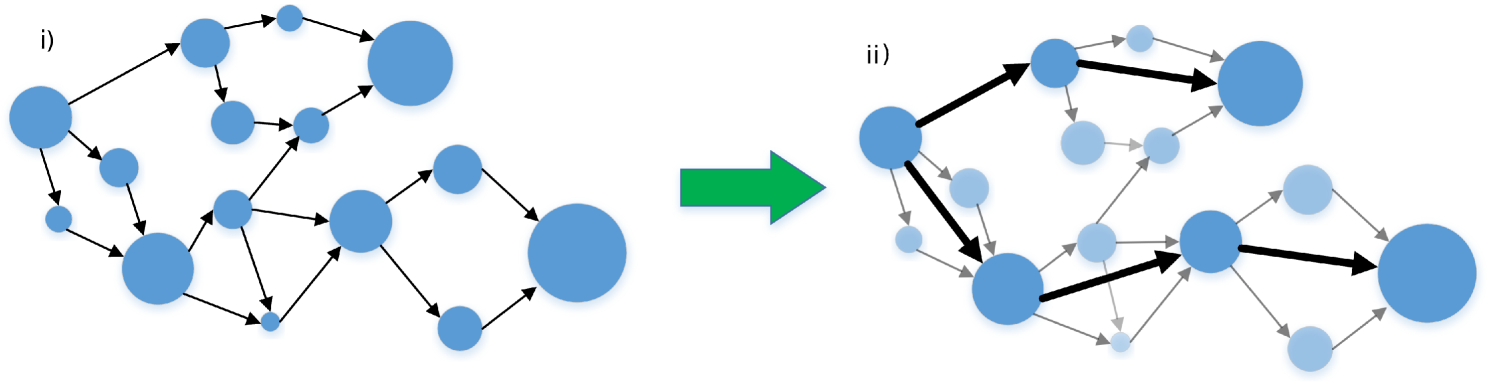
Our algorithm relies on the construction of a skeleton graph (Figure 3(a)) which is a simplified representation of the assembly graph containing only long contigs. The edges in the skeleton graph represent the putative genomic connections of these contigs. As we shall see below, we include an edge in the skeleton graph only if there is sufficient number of long reads that indicate the corresponding adjacency. Since the skeleton graph has a simple structure consisting only of long contigs, mapping long reads to the skeleton graph has less noise and repeat resolution is simpler. Our approach gradually improves the assembly by adding smaller contigs in every iteration.
In each iteration, the algorithm goes through three components (Figure 3(b)): I) Skeleton graph construction/extension; II) Repeat Resolution ; III) Gap bridging.
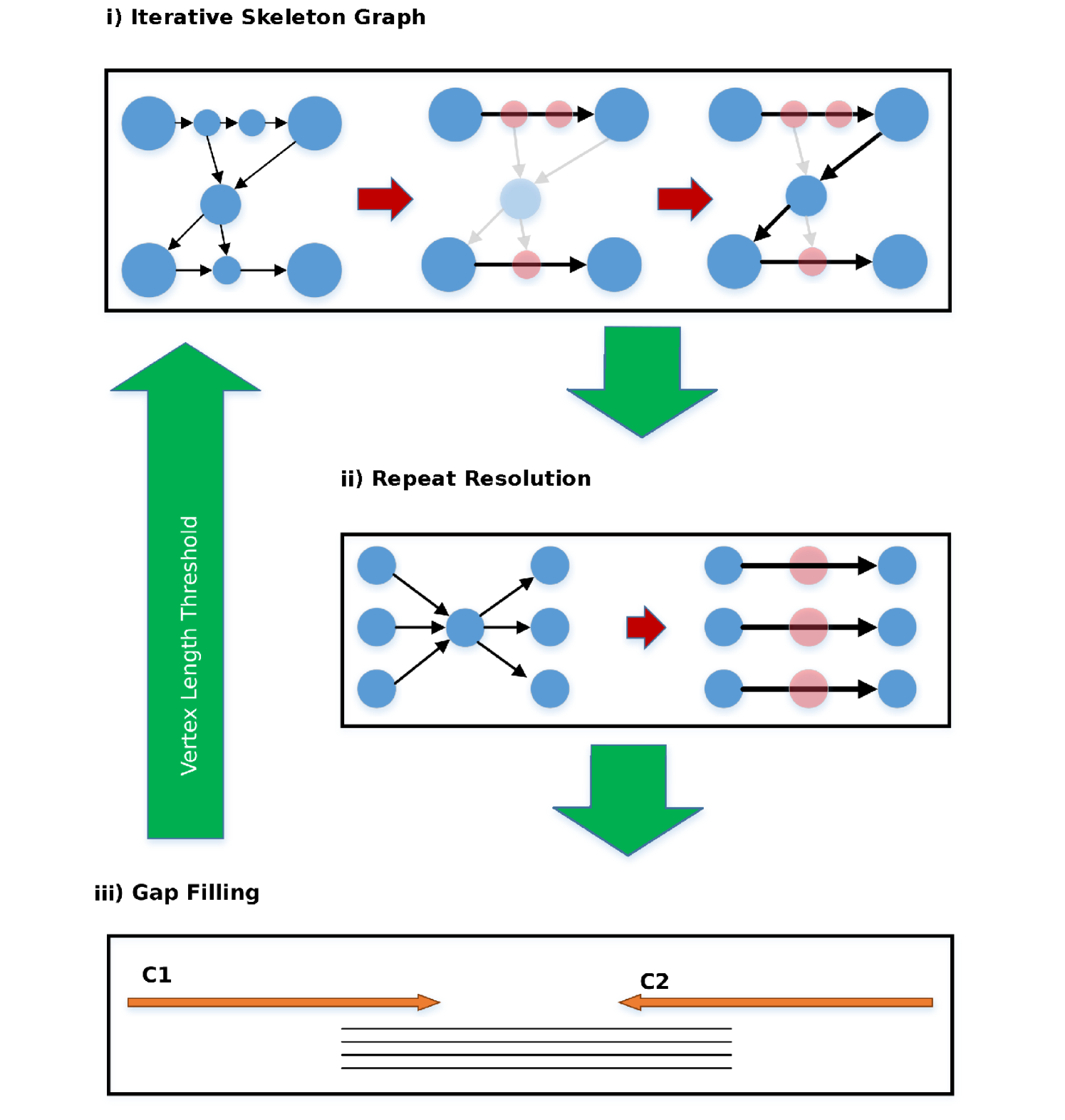
Skeleton graph construction: Given the assembly graph , genome , a length threshold , the skeleton graph has vertex set containing only vertices corresponding to contigs longer than in . An edge depicts a putative adjacent layout between the corresponding contigs in the genome. The skeleton graph represents a simplification of the original assembly graph by ignoring all intermediate short contigs in the assembly graph. The short contigs that occur between consecutive long contigs in the genome are implicitly included by annotating the relevant edges.
Since the skeleton graph is a simpler graph with long vertices, unambiguously mapping the long reads to long contigs and resolving repeats in the skeleton graph is more favorable than in the assembly graph. However, since the genome is unknown, the skeleton graph can not always be constructed in its entirity. Below, we construct an approximate version of skeleton graph using the information from all long read alignments.
The first iteration of the skeleton graph is constructed by using only long contigs from the assembly graph as vertices. Alignments of pairs of long contigs to a long read imply a certain distance (overlap or gap) between the contigs if these were true alignments. A directed edge is added between 2 vertices if there exists a path (or edge) in the assembly graph that certifies the implied distance within certain tolerance. The length of the edge is defined as the distance inferred by following the path (in the assembly graph) rather than the distance inferred from erroneous long reads. In case the adjacent contigs overlap in the assembly graph, then the long reads need to span the entire overlap to include a putative edge between the two contigs in the skeleton graph.
We further refine this approximate graph by the following steps:
Read Count Threshold: We should only keep those edges which have a significant long read support. The length of the long reads is variable. So if we want to resolve a long repeat or fill a large gap, then we will expect a small number of long reads to span the entire repeat or gap. Thus, the expected number of long reads that connect two contigs will depend on the coverage as well as the distance between the two contigs and the read length distribution of the long reads. We thus evaluate the significance of the number of long reads supporting an edge in comparison to support for other competing edges. Our criteria for adding new edges consists of three parts: (i) if the number of long reads supporting an edge is greater than a high confidence threshold that edge is certainly retained; (ii) if the number of supporting long reads is less than a certain low confidence threshold, then such an edge is discarded; (iii) if long read count is between these thresholds, an edge is included either if it is the only outgoing/incoming edge for the source/sink, or if the read count for the edge is significantly higher than other edges incident on source or sink.
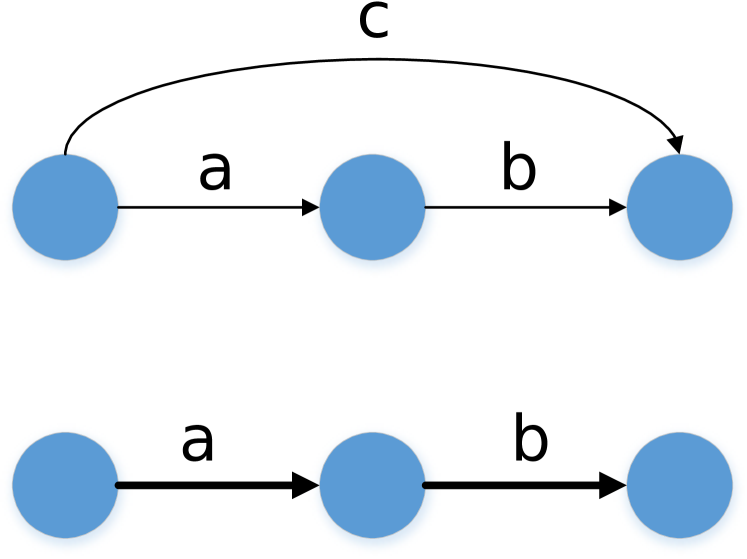
Length-sensitive Transitive Edge Reduction: We can identify the high confidence scaffolds by looking at non-branching paths in the skeleton graph. However, some chains of contigs which ideally should form non-branching paths in the graph may get connected to vertices beyond their immediate neighbours. Such cases can happen when some long reads do not align to the intermediate vertex due to errors. For identification of non-branching paths, we remove the transitive edges which create the false impression of a branch (Figure 4). An edge is defined to be transitive if there is an alternative path from source to sink implying the same distance offset.
Repeat Resolution: The repeat resolution procedure is illustrated in Figure 3(b)(ii). When we remove the transitive edges to identify simple paths, we may lose valuable information from long reads that span repeats appearing as branching nodes in the graph. A branching node will have multiple incoming edges on one side on multiple outgoing edges on the other. The corresponding incoming and outgoing vertices form a bipartite structure as seen in Figure 3(b)(ii). We resolve such repeats by matching pairs of vertices (not necessarily all pairs) in this bipartite structure by looking at reads spanning the repeat. Matched pairs which can be joined unambiguously are then connected by a direct edge (with annotation) and the edges to the intermediate vertex are removed. We do repeat resolution in its simplest form of spanning only one contig at a time. This works if the repeating contig is a maximal repeat. It is the common case for most contigs in our procedure to be maximal repeats since we deal with only long contigs initially. But as we scale to larger genomes with more complex repeat structures of shorter contigs, we need to implement more powerful strategies for spanning repeats.
Gap Bridging: The gap bridging procedure is illustrated in Figure 3(b)(iii). After using all information from connections in the graph to identify the scaffolds, we observe that certain paths terminate as there are no further edges to extend these paths. At this point, we relax the constraint that edges in the skeleton graph should correspond to existing paths of the assembly graph. We can identify possible ways to extend a scaffold by looking at long reads that align from the end of that scaffold to the end of another scaffold. If this is the only possible way to extend either of these scaffolds, then we use these long read alignments to unambiguously bridge the gap between these paths (scaffolds) by adding a new edge between their terminal vertices.
Iteration on Vertex Length: After we have inferred all possible scaffolds from the long contig skeleton, we have the list of all simple paths from this skeleton. Now we can decrease the vertex length threshold according to a vertex length schedule. We add the new short contig vertices (longer than the new lower threshold) either if they are connected to the end vertices of the simple paths from the previous vertex length iteration, or if they are connected to other short vertices. Thus non-terminal non-branching long contigs in scaffolds from one iteration are untouched in the next iteration. Then we iteratively go through the above steps of transitive edge reduction, repeat resolution and gap bridging.
Final assembly: After the completion of all iterations through the vertex length schedule we have our final approximation of the skeleton graph. The simple paths represent our final scaffolds. The edges of these simple paths can either be directed edges in the original assembly graph, or they can be annotated with either path traversals in the assembly graph by the set of long reads bridging a gap. Thus, the final scaffolds can be inferred from these annotated paths on the corresponding sequence of reads that are used for gap filling. Those vertices which are not included in the scaffolds can be inferred as independent contigs.
3 Results
We tested our software for the Escherichia Coli bacterial genome (strain: K12, isolate: MG1665). The short [20] and long [21] read datasets were obtained from the samples provided by Illumina and Pacific Biosciences respectively as described in Table 1.
| Platform | Illumina Hiseq | PacBio RS |
|---|---|---|
| Coverage | 400X | 30x |
| Read Length | 151bpX2 (insert size 300bp) | N50: 5900, Largest:19416 |
| Number of reads | 11 million | 75152 |
Short read assembly: We assembled the short read contigs using the ABySS paired-end assembler with -mer size of 64 base pairs. The computational resources and assembly results are mentioned in Table 2 and Table 3 respectively.
Mapping long reads to contigs: We mapped the long reads to the ABySS assembled contigs using BLASR with minimum percentage identity of 70%.
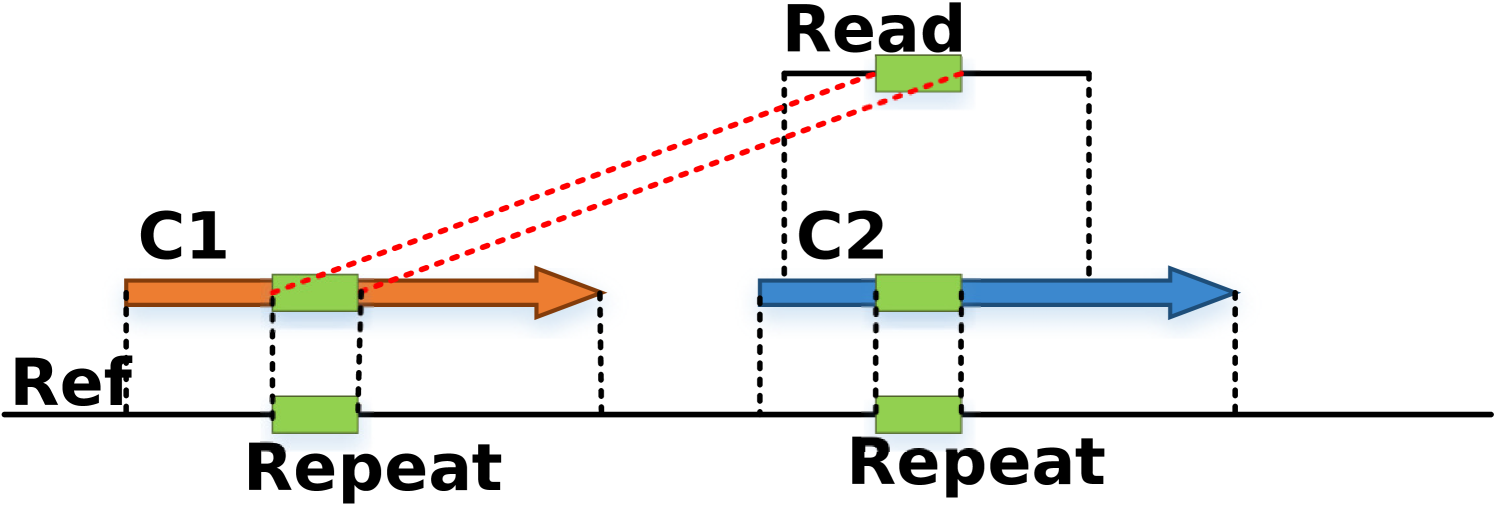
Filtering spurious alignments: There are many short alignments from long reads to contigs due to short repeats contained within contigs as shown in Figure 5. Reads mapping to multiple contigs do not necessarily imply adjacency and need to be filtered. We classify an alignment as long alignment if the unaligned overhang (i.e. length of unaligned portion of the read which ideally should have mapped in case of a true alignment of unerroneous read) is less than 30% of the ideal alignment length (i.e. sum of unaligned overhang and aligned portion). Henceforth, when we refer to alignment of a long read it means it satisfies the criteria for long alignment.
Cerulean scaffolding: We used the ABySS assembly graph and filtered BLASR alignments to generate the scaffolds. The vertex length schedule we used is 2048 bp, 1024 bp, 512 bp, 256 bp and 0 bp.
PacbioToCA: We compare our results to the assembly generated by the alternative approach of assembling error-corrected long reads using PacbioToCA.
AHA scaffolder: We also tested the results of the AHA scaffolder using the ABySS assembled contigs and PacBio long reads as inputs.
ALLPATHS-LG: We did not test ALLPATHS-LG since it requires jumping libraries.
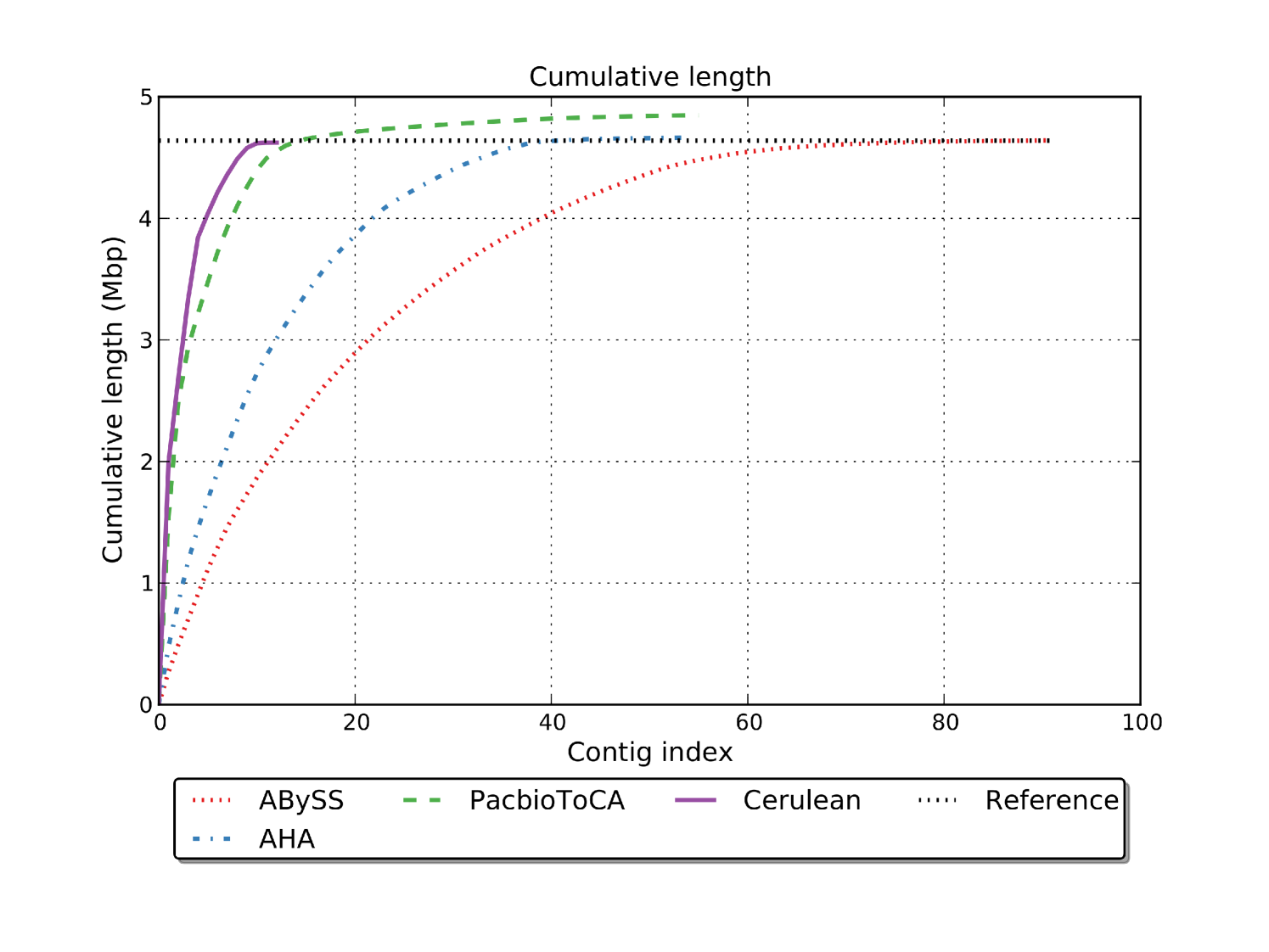
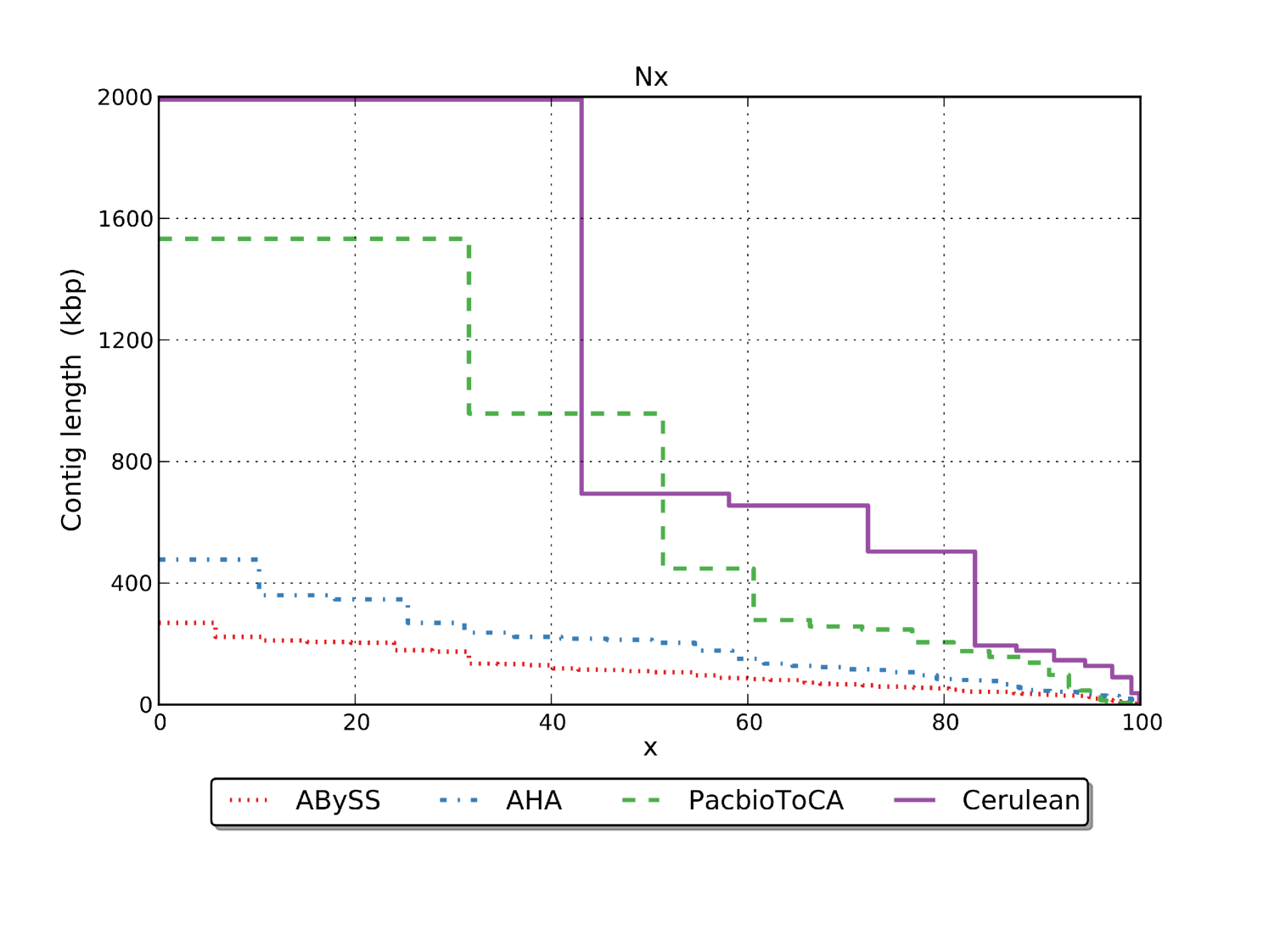
The length distribution comparison of the assembled contigs is displayed in Figures 6(a) and 6(b). We can see that the N50 values for the PacbioToCA assembly is determined solely by the first 2 contigs, but the contig length drops drastically after that giving a very low N75 value of 273 Kbp as compared to N50 of 957 Kbp. The length of the scaffolds generated by Cerulean falls much slower giving a significantly better N75 of 503 Kbp comparable to the N50 length 694 Kbp. Figure 6(a) also shows that the total assembled length for PacbioToCA is significantly larger than the genome length.
| Software | ABySS | BLASR | Cerulean | PacbioToCA | AHA |
|---|---|---|---|---|---|
| Number of threads | 1 | 8 | 1 | 24 | 4 |
| Peak memory usage | 4 GB | 300 MB | 100 MB | 55 GB | 300 MB |
| Runtime | 30 mins | 30 mins | 2 mins | 12 hours | 2 hours |
| Temporary files | 75 MB | 8 MB | 5 MB | 300 GB | 500 MB |
| Software | Reference | ABySS | Cerulean | PacbioToCA | AHA |
|---|---|---|---|---|---|
| # contigs | 1 | 199 | 21 | 55 | 54 |
| # contigs 1000bp | 1 | 83 | 11 | 55 | 48 |
| N50 | 4639675 | 110Kbp | 694KBp | 950Kbp | 213 Kbp |
| N75 | 4639675 | 64KBp | 507KBp | 247 KBp | 107 Kbp |
| Largest contig length | 4639675 | 268969 | 1991897 | 1533073 | 477080 |
| Total length | 4639675 | 4849724 | 4625935 | 4641287 | 4663300 |
| #misassemblies | - | 3 | 4 | 22 | 11 |
Analysis of the final set of contigs indicated that all the 11 long contigs were essentially separated by just 2 non-exact repeats in the reference of lengths (2500 bp and 4300 bp). If we are more aggressive in resolving these repeats, then this approach has the potential to retrieve the entire genome as a single contig. Our conservative adjacency calls did not resolve these repeats in order to retain the accuracy of the assembly. We chose not to make more aggressive decisions in repeat resolution in the dataset because in the case of just 2 repeats, it is easy to implement a scheme that will overfit the data and not scale to other genomes when running in a fully automated setting.
4 Discussions:
Cerulean has a very low resource usage and high accuracy of assembled scaffolds. This makes a very strong case for scaling this approach to larger genomes. The algorithm in its current state focuses on making decisions based on very simple building blocks one at a time. This makes it possible for us to make low risk decisions towards a high accuracy assembly for simple bacterial genomes. However, when analyzing datasets from larger complex genome, we have no prior knowledge of the structure of the repeats and the layout of the contigs generated by short read assemblers. So there are cases where the scaffolding algorithm may not be able to distinguish between a true adjacency signal and a false adjacency signal. In most cases, this will simply stop the algorithm from extending a scaffold due to branching. However, we cannot conclusively rule out the possibility of producing other side effects for every decision made by the algorithm. We also need to acknowledge the fact that we are currently dealing with small bacterial genomes for which we can easily obtain high and more or less uniform coverage for short reads. So far we rely completely on the short read assembler to generate the initial contigs. However, for larger genomes, variable coverage caused due to sequencing bias combined with decisions made by the short read assembler can cause misassembled contigs to start with. In this case, the scaffolder will benefit from not assuming the assembled contigs as ground truth, but actually testing for misassemblies by the short read assembler.
However, the framework of gradual inclusion of complexity does provide us with the opportunity to tackle even more complex genomes in a systematic fashion. Here we discuss a few of these cases where this framework is useful. When extending the scaffolds consisting of large contigs, we aim to extend them unambiguously with the inherent assumption that the larger contigs are usually unique in the reference. We can increase the confidence in this assumption by considering the coverage information of contigs to estimate the repeat counts of contigs and make our decision for extension based on this assumption. Furthermore, if in one iteration of the vertex length schedule, we find an unambiguous way of extending a scaffold, it does not necessarily imply that it is the only way to extend and there can possibly be other ways to extend which require looking at shorter vertices for bridging. One way to address problem is by using the vertex length threshold as a soft cutoff rather than a hard cutoff to allow shorter contigs to compete with the larger contigs if they have very good support from the reads and graph structure.
In Cerulean, we bypass the problems of complex repetitive structures in graph, by only looking at uniquely occurring long vertices. We resolve only one branching vertex at a time under the inherent assumption that most long repeats are maximal repeats isolated from other repeats of comparable length. Larger genomes certainly violate this assumption to a large extent. Thus we may have to connect scaffolds that are separated by multiple branching vertices of comparable lengths. There is also the opportunity to exploit the structure of the graph in extending the scaffolds as shown in Figure 7. A path tracing approach can help us extend scaffolds across such multiple branching vertices. Path tracing is a non-trivial problem in big graphs, but we can use path tracing in the context of our incremental framework to solve specific small problems.
Finally there are techniques we can use to improve our ability to extend scaffolds or add more edges to the skeleton graph. An edge in the skeleton graph can correspond to a walk in the assembly graph. The input mapping from long reads to contigs may miss some alignments from the long reads to intermediate contigs due to high error rate. In such a case, we can perform a more informed search for such a walk by looking at local alignments of reads along neighoring contigs. If we can identify true chains of small contigs, then we can rerun the mapping the reads to the concatenated sequences of these chains and use these mappings while extending existing scaffolds. PBJelly [23] has displayed that gaps in scaffolds can be filled with high accuracy using the mapping reads. Thus while retrieving the the intermediate sequences of scaffolds, we can use a combination of long reads and assembled contigs to bridge the long contigs. After we have finished making the most conservative calls using the assembled short read contigs and filled in all gaps using the pairwise alignments of the long reads, we can be more ambitious and bridge the gaps between the scaffolds with targeted assembly of long reads.
In conclusion, we present a hybrid assembly approach that is both computationally effective and produces high quality assemblies. Our algorithm first operates with a simplified version of the assembly graph consisting only of long contigs and gradually improve the assembly by adding smaller contigs in each iteration. In contrast to the state-of-the-art long reads error correction technique, which requires high computational resources and long running time on a supercomputer even for bacterial genome datasets, our software can produce comparable assembly using only a standard desktop in a short running time.
Acknowledgments
The authors will like to sincerely thank Pavel Pevzner and Glenn Tesler for their insightful comments. V.D. and V.B. were supported in part by NIH grants 5RO1-HG004962 and U54 HL108460, and by the NSF grant NSF-CCF-1115206. S.P. was supported in part by NIH grant 3P41RR024851-02S1.
Disclosure Statement
No competing financial interests exist.
References
- [1] Koren, S., Schatz, M.C., Walenz, B.P., Martin, J., Howard, J.T., Ganapathy, G., Wang, Z., Rasko, D.A., McCombie, W.R., Jarvis, E.D., et al.: Hybrid error correction and de novo assembly of single-molecule sequencing reads. Nature biotechnology 30(7) (2012) 693–700
- [2] Staden, R.: A strategy of dna sequencing employing computer programs. Nucleic acids research 6(7) (1979) 2601–2610
- [3] Myers, E.W.: The fragment assembly string graph. Bioinformatics 21(suppl 2) (2005) ii79–ii85
- [4] Myers, E.W., Sutton, G.G., Delcher, A.L., Dew, I.M., Fasulo, D.P., Flanigan, M.J., Kravitz, S.A., Mobarry, C.M., Reinert, K.H., Remington, K.A., et al.: A whole-genome assembly of drosophila. Science 287(5461) (2000) 2196–2204
- [5] Simpson, J.T., Durbin, R.: Efficient de novo assembly of large genomes using compressed data structures. Genome Research 22(3) (2012) 549–556
- [6] Idury, R.M., Waterman, M.S.: A new algorithm for dna sequence assembly. Journal of Computational Biology 2(2) (1995) 291–306
- [7] Pevzner, P.A., Tang, H., Waterman, M.S.: An eulerian path approach to dna fragment assembly. Proceedings of the National Academy of Sciences 98(17) (2001) 9748–9753
- [8] Chaisson, M.J., Pevzner, P.A.: Short read fragment assembly of bacterial genomes. Genome research 18(2) (2008) 324–330
- [9] Simpson, J.T., Wong, K., Jackman, S.D., Schein, J.E., Jones, S.J., Birol, İ.: Abyss: a parallel assembler for short read sequence data. Genome research 19(6) (2009) 1117–1123
- [10] Zerbino, D.R., Birney, E.: Velvet: algorithms for de novo short read assembly using de bruijn graphs. Genome research 18(5) (2008) 821–829
- [11] Eisenstein, M.: Companies’ going long’generate sequencing buzz at marco island. Nature biotechnology 31(4) (2013) 265–266
- [12] Waldbieser, G.: Production of long (1.5 kb–15.0 kb), accurate, dna sequencing reads using an illumina hiseq2000 to support de novo assembly of the blue catfish genome. In: Plant and Animal Genome XXI Conference, Plant and Animal Genome (2013)
- [13] Chin, C.S., Alexander, D.H., Marks, P., Klammer, A.A., Drake, J., Heiner, C., Clum, A., Copeland, A., Huddleston, J., Eichler, E.E., et al.: Nonhybrid, finished microbial genome assemblies from long-read smrt sequencing data. Nature methods (2013)
- [14] Au, K.F., Underwood, J.G., Lee, L., Wong, W.H.: Improving pacbio long read accuracy by short read alignment. PLoS One 7(10) (2012) e46679
- [15] Hercus, C.: Novocraft short read alignment package. Website http://www. novocraft. com (2009)
- [16] Wu, T.D., Watanabe, C.K.: Gmap: a genomic mapping and alignment program for mrna and est sequences. Bioinformatics 21(9) (2005) 1859–1875
- [17] Bashir, A., Klammer, A.A., Robins, W.P., Chin, C.S., Webster, D., Paxinos, E., Hsu, D., Ashby, M., Wang, S., Peluso, P., et al.: A hybrid approach for the automated finishing of bacterial genomes. Nature biotechnology (2012)
- [18] Ribeiro, F.J., Przybylski, D., Yin, S., Sharpe, T., Gnerre, S., Abouelleil, A., Berlin, A.M., Montmayeur, A., Shea, T.P., Walker, B.J., et al.: Finished bacterial genomes from shotgun sequence data. Genome research 22(11) (2012) 2270–2277
- [19] Chaisson, M.J., Tesler, G.: Mapping single molecule sequencing reads using basic local alignment with successive refinement (blasr): application and theory. BMC bioinformatics 13(1) (2012) 238
- [20] : E.Coli MG1655 Illumina HiSeq2000 sequencing dataset. ftp://webdata:webdata@ussd-ftp.illumina.com/Data/SequencingRuns/MG1655/MiSeq_Ecoli_MG1655_110721_PF.bam (2013) [Online; accessed 24-June-2013].
- [21] : E.Coli K12 MG1655 Pacbio RS sequencing dataset. http://files.pacb.com/datasets/primary-analysis/e-coli-k12/1.3.0/e-coli-k12-mg1655-raw-reads-1.3.0.tgz (2013) [Online; accessed 24-June-2013].
- [22] Schmutz, J., Wheeler, J., Grimwood, J., Dickson, M., Yang, J., Caoile, C., Bajorek, E., Black, S., Chan, Y.M., Denys, M., et al.: Quality assessment of the human genome sequence. Nature 429(6990) (2004) 365–368
- [23] English, A.C., Richards, S., Han, Y., Wang, M., Vee, V., Qu, J., Qin, X., Muzny, D.M., Reid, J.G., Worley, K.C., et al.: Mind the gap: Upgrading genomes with pacific biosciences rs long-read sequencing technology. PloS one 7(11) (2012) e47768