Pre-training Co-evolutionary Protein Representation via A Pairwise Masked Language Model
Abstract
Understanding protein sequences is vital and urgent for biology, healthcare, and medicine. Labeling approaches are expensive yet time-consuming, while the amount of unlabeled data is increasing quite faster than that of the labeled data due to low-cost, high-throughput sequencing methods. In order to extract knowledge from these unlabeled data, representation learning is of significant value for protein-related tasks and has great potential for helping us learn more about protein functions and structures. The key problem in the protein sequence representation learning is to capture the co-evolutionary information reflected by the inter-residue co-variation in the sequences. Instead of leveraging multiple sequence alignment as is usually done, we propose a novel method to capture this information directly by pre-training via a dedicated language model, i.e., Pairwise Masked Language Model (PMLM). In a conventional masked language model, the masked tokens (i.e. amino acid residues) are modeled by conditioning on the unmasked tokens only, but processed independently to each other. However, our proposed PMLM takes the dependency among masked tokens into consideration, i.e., the probability of a token pair is not equal to the product of the probability of the two tokens. By applying this model, the pre-trained encoder is able to generate a better representation for protein sequences. Our result shows that the proposed method can effectively capture the inter-residue correlations and improves the performance of contact prediction by up to compared to the MLM baseline under the same setting. The proposed model also significantly outperforms the MSA baseline by more than on the TAPE contact prediction benchmark when pre-trained on a subset of the sequence database which the MSA is generated from, revealing the potential of the sequence pre-training method to surpass MSA based methods in general.
1 Introduction
Life is ruled by biological sequences and molecules, i.e. DNA, RNA, and protein sequences, following the de facto ‘natural’ language of biology. For protein, the structure is determined by the sequence, and its function is realized by the structure, according to Anfinsen’s dogma. However, the structure and function label is never easy to obtain (time-consuming and expensive) nor always effective due to the dark regions and structure dynamics. On the other hand, the number of unlabeled protein sequences increases quite faster than that of the labeled ones, due to the large gap between low-cost, high-throughput sequencing methods and expensive yet time-consuming labeling approaches by time- and labor-intensive manual curation process (Consortium, 2019). Understanding the protein sequences is vital to advance in biology, healthcare, and medicine. The protein sequence representation has been exploited in various real applications, including for dark proteome (Perdigão et al., 2015), where the ‘dark’ regions of proteins are never observed by experimental structure determination and inaccessible to homology modeling; therapies, such as cancer diagnosis (Vazquez et al., 2009) and antibody design (Whitehead et al., 2012); function/property prediction for protein engineering, such as fitness prediction (Hsu et al., 2021); phylogenetic inference (Hie et al., 2021b); sequence mutations, such as learning mutational semantics (Hie et al., 2020), virus evolution and escape prediction (Hie et al., 2021a), and cancer diagnosis by mutation prediction (Reva et al., 2011; Martelotto et al., 2014), to name a few.
Co-evolutionary information represented by the inter-residue co-variation is essential for protein sequences in terms of both structure and function due to the evolution pressure from natural selection – sequences with more stable structure and more adequate function are usually remained. A representation that can capture this information from the sequences is beneficial for understanding the molecular machinery of life. This information is previously quantified by analyzing homologous sequences, a.k.a multiple sequence alignment (MSA), for the target sequence, where the homologs are retrieved from public protein sequence databases by applying curated procedure and intensive computation with customized tools and hyper-parameters. For protein function prediction, unsupervised models can be learned from these homologous sequences, such as mutational effect prediction from sequence co-variation (Hopf et al., 2017) and mutational landscape inference (Figliuzzi et al., 2016). It is demonstrated that incorporating inter-residue dependencies using a pairwise model that can power the predictions to agree more with the mutational experiment observations (Mann et al., 2014; Boucher et al., 2016; Riesselman et al., 2017). The reason is that, the function of the sequence is the combination effect of the residues other than the sum of the properties of each residue (Hopf et al., 2017). As the cost to quantify the co-effect of multiple residues is combinatorial while modeling their interactions is critical, pairwise co-variation analysis becomes the best choice, meanwhile, Figliuzzi et al. (2018) demonstrates that pairwise models are able to capture collective residue variability. For protein structure prediction, the key step is to predict inter-residue contacts/distances, while the shared cornerstone of prediction is performing evolutionary coupling analysis, i.e. residue co-evolution analysis, on the constructed MSA for a target protein (Ju et al., 2021). The underlying rational is that two residues which are spatially close in the three-dimensional structure tend to co-evolve, which in turns can be exploited to estimate contacts/distances between residues (Seemayer et al., 2014; Jones & Kandathil, 2018). Although the contacts are usually rare in the residue pairs of the sequence, they are critical to rebuilding the 3D structure of the protein due to their roles of constraints on the sketch.
A natural question arises that, as both derived from unlabeled sequences, can we directly capture this co-evolutionary information via sequence pre-training instead of extracting from MSA? To answer this question, we first discuss the key ingredient missed by conventional language models, then we propose a pairwise masked language model for the protein sequence representation learning. The key information to extract from the MSA (denoted as ) is the statistics for , typically the co-variation for each pair, where are two indexes of the tokens/residues in the sequences. The intuition is that, if the two residues are independent, their co-variance will be close to zero, otherwise, it indicates the co-evolutionary relation between these two positions.
| - | ||||
| - | ||||
| - | - | |||
| 9% | ||||
While for the conventional masked language model, although they scan through all the same sequences as MSA-based methods and the losses for all the residues in the masked tokens will be back-propagated, they are processed in an accumulative way within the batches, i.e. independently updating the weights. This means that is modeled by the LMs. Table 1 presents an example for this kind of method, originating from NLP but sub-optimal for protein sequences due to the critical and much frequent co-evolution relationship among the residues. Here, we argue that for the protein sequences . Auto-regressive methods, i.e. generative pre-training (GPT), where the latter tokens are predicted by conditioning on the previous ones from the same direction, still having the similar issue. Inspired by this, we design a pairwise masked language model and pre-train the model with a pairwise loss calculated from the protein sequences directly. Following the masked language model where the tokens of each sequence are picked at a probability and then be masked, replaced, or kept, our proposed model takes the masked sequences as input and predicts these original tokens, especially, the label for each masked token pair.
To examine the capability of the model to capture the co-evolutionary information, we select protein contact prediction as our main downstream task due to its well-established relationship to the co-evolutionary information and the plentiful data that are publicly available and well measured by empirical experiment. Here, protein contact prediction is a binary classification task for amino acid residue pairs, a residue pair is called a contact if their distance is less than or equal to a distance threshold, typically (i.e., ). The result shows that this method can significantly improve the learned representation on the contact prediction task, it even surpasses the MSA baseline when pre-trained on another sequence subset UR50 with the sequence-only input on the TAPE benchmark. Meanwhile, an additional experimental evaluation for secondary structure prediction illustrates that the proposed model does not hurt performance, while significant improvements are observed in some other settings for remote homology prediction.
2 Related Work
Evolutionary Coupling Analysis
Co-evolution information is closely correlated to the contacts, due to the rational that two residues are likely co-evolving when they are spatially close. To this end, techniques such as mutual information (MI) (Gloor et al., 2005) are exploited to quantify this feature. Later than MI, direct coupling analysis (DCA) proves to be more accurate and be widely adopted, for example, EVfold (Sheridan et al., 2015), PSICOV (Jones et al., 2012), GREMLIN (Kamisetty et al., 2013), plmDCA (Ekeberg et al., 2013), CCMpred (Seemayer et al., 2014) are all built on DCA. For protein sequences, the common idea of these methods is to use statistical modeling to quantify the strength of the direct relationship between two positions of a protein sequence, excluding effects from other positions. A variety of DCA methods are further proposed to generate the direct couplings of residues by fitting Potts models (Ekeberg et al., 2013) or precision matrix (Jones et al., 2012) to MSAs, e.g. mean-field DCA (Morcos et al., 2011), sparse inverse covariance (Jones et al., 2012) and pseudo-likelihood maximization (Ekeberg et al., 2013; Balakrishnan et al., 2011; Seemayer et al., 2014).
By taking DCA-derived scores as features, deep neural networks based supervised methods significantly outperform the unsupervised methods (Senior et al., 2020; Jones & Kandathil, 2018; Wang et al., 2017; Xu, 2019; Yang et al., 2020). Most neural network models, including AlphaFold (AlQuraishi, 2019) and RaptorX (Xu, 2019), rely on this feature. However, due to the considerable information loss after transforming MSAs into hand-crafted features, supervised models, such as CopulaNet (Ju et al., 2021) and AlphaFold2 (Jumper et al., 2021), are proposed to directly build on the raw MSA. The superior performance over the baselines demonstrates that residue co-evolution information can be mined from the raw sequences by the model. A noticeable drawback of the MSA based methods is that, when the MSA quality (i.e. the number of the homologous sequences) is low for a target sequence, the model performance drops a lot.
Pre-Training Methods
Following the pre-train and fine-tune paradigm (Peters et al., 2018; Kenton & Toutanova, 2019; Liu et al., 2019; Radford et al., 2018), various pre-training methods are proposed recently to learn better representations for protein sequences. TAPE (Rao et al., 2019) is built as a benchmark to evaluate the protein sequence pre-training method. It demonstrates the performance can be improved by pre-training compared to one-hot representation, however, also indicates that the performance of the pre-training based models on pure sequences still lags behind the alignment-based method in the downstream tasks.
Among pre-training methods, RNNs are exploited as the pre-training model. Bepler & Berger (2019) use two layers of Bi-LSTM as an extraction part of the original protein sequences by applying next token prediction for pre-training language modeling. UniRep (Alley et al., 2019) uses a similar training scheme as Bepler & Berger (2019), then use evo-turning (evolutionary fine-tuning) to address the importance of evolutionary information for the protein embedding. UDSMProt (Strodthoff et al., 2020) relies on an AWD-LSTM language model, which is a three-layer LSTM regularized by different kinds of dropouts.
A variety of techniques based on Transformer (Vaswani et al., 2017) are applied to build dedicated models for proteins. ESM (Rives et al., 2021) demonstrates that Transformer models can outperform RNN based models, it also illustrates that the protein sequence diversity and model size have significant impacts on the pre-trained encoder performance. PRoBERTa (Nambiar et al., 2020) follows RoBERTa to pre-train the model with Byte Pair Encoding and other optimization techniques. Lu et al. (2020) applies contrastive learning by mutual information maximization to the pre-training procedure, MSA Transformer (Sturmfels et al., 2020) learns the representation on the MSAs for a protein sequence, however, it relies on expensive database searches to generate the required MSAs for each sequence.
Large-scale models are explored due to the fact that protein sequence datasets are large in size and complex in interaction. ProtTrans (Elnaggar et al., 2020) trains the protein LMs (pLMs) on the Summit supercomputer using more than thousand GPUs and TPU Pod up-to cores, the most informative embeddings even outperform state-of-the-art models without multiple sequence alignments (MSAs) for the secondary structure prediction. Xiao et al. (2021) also demonstrate that sequence evolution information can be accurately captured by a large-scale model from pre-training, up to billion parameters pre-trained on a GPUs cluster.
Additional supervised labels are exploited for protein sequence pre-training. PLUS (Min et al., 2019) tries to model the protein sequence with masked language model together with an auxiliary task, i.e. same family prediction, in their work. Sturmfels et al. (2020) add a pre-training task named profile prediction for pre-training.
Generative pre-training is also exploited for protein engineering, for example, ProGen (Madani et al., 2020) is a generative model conditioned on taxonomic information as an unsupervised sequence generation problem in order to leverage the exponentially growing set of proteins that lack costly, structural annotations.
The outputs of the pre-trained language models can be used in different ways. For example, Vig et al. (2020), Rao et al. (2020), and Bhattacharya et al. (2020) demonstrate that the attention weights from the pre-trained models have a strong correlation with the residue contacts. Hie et al. (2021b) analyze the correlation between the pseudo likelihood of the mutated sequences predicted by the language model and the evolution of the mutations, the results suggests that pre-trained language models on protein sequences are able to predict evolutionary order at different timescales. Hie et al. (2021a; 2020) apply pre-trained protein language models to predict mutational effects and virus escape.
3 Method
Given the sequence data , where , language models can be built on the sequences for pre-training.
3.1 Pairwise Masked Language Model
The loss function of vanilla Masked Language Model (MLM) can be formulated as follows:
where is the sequence set, is a sequence in , represents the masked sequence of where the masked token indices are in , stands for the -th token in the sequence , and denotes the encoder parameters.
In this paper, we propose a novel Pairwise Masked Language Model (PMLM) whose loss function can be written as:
The combined loss for both MLM and PMLM can be used for pre-training as:
where is a weight coefficient to balance two losses. In this paper, is set to during pre-training the PMLM models. As we can see, all the MLM and PMLM labels for pre-training are from the sequences themselves, thus still self-supervised. For multiple rounds of updates, if -th and -th positions are independent, degenerates to . When they are co-evolutionary, the model learns a different distribution from the independent case.
3.2 Model Architecture
The goal of pre-training is to build a good protein sequence encoder that generates better representation. The pre-training model mainly consists of a protein sequence encoder and two prediction heads, i.e., a token prediction head and a pair prediction head. The sequence encoder is built on stacked Transformer encoder layers. The overview of our model is illustrated in Figure 1. Transformer is believed to be a powerful tool for modeling sequence data and has been applied to various tasks, including natural language understanding, question answering, computer vision, and so on. Thus we exploit a Transformer encoder as the sequence encoder of our model. The sequence encoder takes raw protein sequences as input and converts them into their vector representations. The model was trained on protein sequences using both masked token prediction (MLM) and masked pair prediction (PMLM) for protein language modeling. Each prediction head is a MLP, i.e. two-layer fully-connected (FC) neural network, where the output is mapped into the vocabulary via softmax, i.e. amino acid residues (denoted as ) for the token prediction head and amino acid pairs (denoted as ) for the pair prediction head.

3.3 Masked Pair Prediction
The masked language model lets the model to reconstruct the masked tokens conditional on the other tokens in the sequences. Predicting masked pairs follows the same idea of the masked token prediction, however, with losses for the pairs. A two-layer FC neural network is exploited for the prediction of masked pairs, whereas another one for that of masked tokens. During pre-training, the dot and difference of the vectors for the residue pair are concatenated before feeding into the pair prediction head. Then, the output is mapped into a vector of dimension by softmax. This prediction is finally compared with the pair label from the sequence itself.
It is not trivial to generate pair labels from multiple masked tokens. The pairwise label construction process is demonstrated in Figure 2. For each pair for the masked token where , a pairwise label is generated as , where and . Obviously, we have for and . The pairs for whose are ignored since they are already a part of the MLM loss. Notably, does not necessarily mean since two different positions may be the same residue. For each valid pair, the pair prediction head generates the probability over . For a masked sequence , the size of the pairwise labels is while each label is a one-hot encoding for .
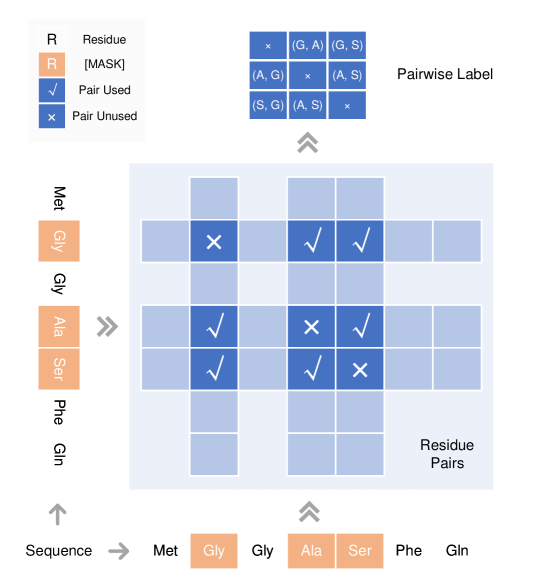
Intuitively, the masked pair loss is consistent with the masked token loss. For example, the loss can be treated as a combined loss for the two masked tokens if the two positions within the pair are independent, otherwise this masked pair loss provides extra information of the sequence besides each position.
3.4 Fine-tuning for Contact Prediction
The generated encoder is further fine-tuned on a given supervised dataset for contact prediction. As the prevalent approach does, different neural networks can be built on top of the pre-trained model for further fine-tuning, whereas, a simple FC layer followed by a softmax operator can be applied to evaluate the representation. For the residue pairs task, e.g. contact prediction, the fine-tuning network is built on top of the first FC layer of the pair prediction head, other tasks are fine-tuned on the encoder outputs.
4 Experiment Evaluation
4.1 Experiment Setup
To evaluate the performance of the pairwise masked language model, several models with different settings are pre-trained on two prevalent datasets, i.e. Pfam (El-Gebali et al., 2019) and UR50 (release 2018_03). Following the RoBERTa-base setting, the hidden size, feed forward dimension, number of encoder layers, and attention heads of the base models are set as , , , respectively (denoted as MLM-base for MLM and PMLM-base for PMLM). A larger model named PMLM-large is pre-trained with the same setting except using as the number of encoder layers. Moreover, the largest model with a hidden size of and a number of encoder layers of is also pre-trained on UR50, denoted as PMLM-xl. The implementation is optimized to speed up the training procedure, e.g., the pairwise loss is applied alone by enabling the diagonal elements and disabling the token prediction head. MLM-base and PMLM-base are pre-trained with maximum length , while PMLM-large is pre-trained with a maximum length of . The positional encoding of both models are non-learnable. MLM-base and PMLM-base are pre-trained using the Adam optimizer with peak learning rate and clip norm , the learning rate scheduler is a polynomial decay scheduler where the learning rate is decreased linearly after warming up by steps to the peak. The hyper-parameters are almost the same for pre-training PMLM-large except that the peak learning rate is set to . MLM-base, PMLM-base, and PMLM-large are pre-trained on Tesla V100 GPU cards, about three weeks for MLM-base/PMLM-base and about seven weeks for PMLM-large. PMLM-xl is pre-trained on Tesla V100 GPU cards for more than two weeks.
Two groups of experiments are conducted to examine the generated model on the TAPE contact prediction test set (denoted as TAPE-Contact) and the RaptorX contact prediction test set (denoted as RaptorX-Contact). The models are all evaluated on or fine-tuned from the checkpoint of the sixtieth epoch, except PMLM-base on UR50, which is fine-tuned from that of the ninetieth epoch. For TAPE-Contact, we use a shallow decoder as TAPE does, i.e. simply use an outer-dot and outer-difference of each pair followed by a linear projection with softmax, for the sequence representation to evaluate the representation. To see the potential of the representations, RaptorX-Contact is evaluated on a more expressive decoder, i.e., a ResNet with stacked convolution blocks, on top of the pre-trained encoder as ESM does, however, the convolution blocks are not dilated. The fine-tuning process for each model on each dataset is finished on one Tesla V100 GPU card.
4.2 Pre-training Validation
The two heads of the PMLM model output and separately, this raises another question that if they have already learned the difference between the independent and dependent positions of the sequence. To answer this question, we aggregate the validation losses and accuracy scores of pre-training on the Pfam and UR50 sequence datasets, as shown in Table 2. As we can see, even the fine-tuned performance of PMLM-base is better than that of MLM-base, the loss of PMLM-base is slightly higher than that of MLM-base. We further use to quantify the difference. For PMLM-base and PMLM-large on Pfam and UR50, is not negligible as an expectation, which indicates the difference of the correct probability for the most likely residue on each position. The joint probability is quite different to the multiplication of the individual probability, illustrating that PMLM is able to capture the co-evolutionary information via pre-training on pure sequences. This observation can be further validated by the histograms of of the PMLM-large model on the Pfam and UR50 validation sequences, which is shown in Figure 3, each pair of the sequence is masked when predicting the probabilities.
| Model | Data | Accmlm | Accpmlm | Acc | Acc / Accpmlm | ||
|---|---|---|---|---|---|---|---|
| MLM-base | Pfam | 1.6475 | - | 48.0% | - | - | - |
| PMLM-base | Pfam | 1.6728 | 3.3446 | 47.1% | 22.4% | 0.22% | 1.0% |
| MLM-base | UR50 | 2.2725 | - | 32.0% | - | - | - |
| PMLM-base | UR50 | 2.2784 | 4.5557 | 31.8% | 10.9% | 0.79% | 7.2% |
| PMLM-large | UR50 | 2.1710 | 4.3419 | 35.1% | 13.2% | 0.88% | 6.7% |
| PMLM-xl | UR50 | - | 3.9746 | 37.3% | 16.9% | 2.95% | 17.5% |


4.3 Comparison on TAPE-Contact
For a fair comparison, the models are evaluated on the TAPE-Contact with the same contact prediction head, which is an FC layer with a dropout followed by a softmax operator. Medium and long range precision at is used for evaluation, where medium and long range means the there are at least residues between the pair to test. With precision at , the top contact predictions are compared to the ground-truth, where is the length of the input sequence.
To better control the setting, a Transformer encoder for MLM (denoted as MLM-base) as well as a Transformer encoder (denoted as PMLM-base) are pre-trained on the Pfam and UR50 datasets. The hyper-parameters for these models resemble that of the RoBERTa-base for NLP, i.e., hidden size as , feed forward dimension as , and number of layers as . To further evaluate the effect of PMLM, we also pre-train a deeper Transformer encoder with the number of layers increased to , denoted as PMLM-large.
The fine-tuned performance of the models are compared in Table 3. The performance of TAPE transformer and MSA baseline are reported in the paper. As we can see, the performance is increased from for TAPE to for MLM-base. Comparing the two models which is close in model size pre-trained on Pfam, i.e. MLM-base and PMLM-base, a huge performance gain over is witnessed, demonstrating the positive effect of the PMLM for pre-training.
Notably, PMLM-xl pre-trained on UR50 can even outperform the MSA baseline (about ), although UR50 is a small subset of UniParc + Metagenome, which shields the light for the single sequence pre-training methods to exceed the MSA based methods.
| Model | #Params | Data | Pre-train Task | Result |
|---|---|---|---|---|
| TAPE Transformer | 38M | Pfam | MLM | 36.0 |
| MLM-base | 85M | Pfam | MLM | 37.3 |
| PMLM-base (ours) | 87M | Pfam | MLM + PMLM | 47.2 |
| PMLM-base (ours) | 87M | UR50 | MLM + PMLM | 57.7 |
| PMLM-large (ours) | 250M | UR50 | MLM + PMLM | 66.7 |
| PMLM-xl (ours) | 713M | UR50 | MLM + PMLM | 71.7 |
| MSA baseline | - | UniParc + Metagenome | - | 64.0 |
4.4 Comparison on RaptorX-Contact
The models are evaluated on the RaptorX-Contact with the same contact prediction module, however, with different contact prediction modules, customized networks may be optimal for different pre-trained encoder.
As shown in Table 4, PMLM-base outperforms ESM Transformer-12 by about given that their model sizes are close, demonstrating the effect of PMLM for pre-training again. When comparing the larger models of a close model size pre-trained, PMLM-xl significantly outperforms ESM models (i.e., more than for ESM Transformer-34 and for ESM-1b) even that a systematic hyper-parameter searching on a 100M model is conducted to generate the hyper-parameters for ESM-1b, which is not performed for PMLM-xl, showing the superior performance of the PMLM encoder.
| Model | #Params | Data | Pre-train Task | Result |
|---|---|---|---|---|
| TAPE Transformer | 38M | Pfam | MLM | 23.2 |
| UniRep | 18M | UR50 | Autoregressive | 21.9 |
| SeqVec | 93M | UR50 | Autoregressive | 29.0 |
| ESM Transformer-12 | 85M | UR50 | MLM | 37.7 |
| PMLM-base (ours) | 87M | UR50 | MLM + PMLM | 41.6 |
| ESM Transformer-34 | 669M | UR50 | MLM | 50.2 |
| ESM-1b | 650M | UR50 | MLM | 56.9 |
| PMLM-xl (ours) | 715M | UR50 | MLM + PMLM | 59.9∗ |
4.5 Ablation Study
To study the factors which influence the model performance, a systemic ablation study is conducted.
Pre-training Data
Pfam is a dataset with protein sequences from a few thousand families, while UR50 consists of much more diverse sequences, i.e., sequences from UniRef with 50% sequence identity. In other words, UR50 is more diverse than Pfam and representative for more sequences. As shown in Table 3, when PMLM-base is pre-trained on UR50, which consists of sequences with higher diversity, instead of Pfam, the performance gain for the task is more than , increased from to . As shown in Table 5,
| Model | #Params | Data | Pre-train Task | Result |
|---|---|---|---|---|
| MLM-base (+ Linear) | 85M | Pfam | MLM | 19.8 |
| PMLM-base (+ Linear) | 85M | Pfam | MLM + PMLM | 25.2 |
| MLM-base (+ Linear) | 85M | UR50 | MLM | 36.9 |
| PMLM-base (+ Linear) | 87M | UR50 | MLM + PMLM | 39.2 |
| PMLM-base (+ ResNet) | 88M | UR50 | MLM + PMLM | 41.6 |
| PMLM-large (+ ResNet) | 252M | UR50 | MLM + PMLM | 54.1 |
| PMLM-xl (+ ResNet) | 715M | UR50 | MLM + PMLM | 59.9 |
Model Size
Model under-fitting is observed in both ESM model pre-training and ours. A straightforward way to tackle this is by increasing the model size. To examine this factor, we compare two models with different sizes pre-trained on the same data, i.e. PMLM-base and PMLM-large. As shown in Table 3, when PMLM-base and PMLM-large are both pre-trained on UR50, the performance gap is about between (PMLM-base) and (PMLM-xl), illustrating that increasing model size indeed benefits the performance for contact prediction. Performance increases by more than as shown in Table 5 when comparing PMLM-xl (+ ResNet) with PMLM-base (+ ResNet). Both are pre-trained on UR50.
Downstream Task Module
On top of the pre-trained encoder, there are various methods to fine-tune the model for downstream tasks. To study the impact of this factor, we also compare the pre-trained encoder PMLM-base with two different downstream modules, namely, a simple linear layer (+ Linear) and an eight-layer ResNet with convolution blocks (+ ResNet). As shown in Table 4, by comparing the performance of PMLM-base (+ Linear) and PMLM-base (+ ResNet), we can see that the precision score is increased from to , indicating that a more expressive model can be trained to improve the performance on the ever pre-trained encoder.
Other Downstream Tasks
| Model | CB513 | CASP12 | TS115 |
|---|---|---|---|
| TAPE Transformer | 0.730 | 0.710 | 0.770 |
| PMLM-base (pre-trained on Pfam, ours) | 0.728 | 0.706 | 0.771 |
| Model | Fold | Superfamily | Family |
|---|---|---|---|
| TAPE Transformer | 0.210 | 0.340 | 0.880 |
| PMLM-base (pre-trained on Pfam, ours) | 0.199 | 0.446 | 0.946 |
To evaluate our pre-trained encoder on other tasks, we also compared the performance of PMLM-base and the TAPE Transformer for Secondary Structure Prediction and Remote Homology Prediction on the TAPE benchmark, the results in terms of accuracy scores are listed in Tables 6 and 7. The performance numbers of PMLM-base for secondary structure prediction are quite close to that of the TAPE baseline Transformer, illustrating that the proposed model does not hurt the performance with the additional loss. For Remote Homology Prediction, the performance gap varies on the fold-level, superfamily-level, and family-level holdout test sets. The performance of PMLM-base is slightly worse than that of the TAPE Transformer on the fold-level test set, however, much better on the superfamily-level and family-level test sets up to 10%, showing the potential improvement from the extracted co-evolutionary information on other tasks.
5 Discussion
In this paper, we propose a novel model named pairwise masked language model for the protein sequence encoder to capture co-evolutionary information during pre-training. The pre-trained model generates a better representation for global structure by applying this model. Our result shows that the proposed method significantly improves the performance of contact prediction compared to the baselines. Meanwhile, the proposed model surpasses the MSA baseline on the TAPE contact benchmark. Although the performance is encouraging, the potential capability of the proposed model, i.e. PMLM, is not fully developed, neither it is the only way to extract the co-evolutionary information from the sequences. For example, an observation is that increasing the mask probability for the tokens might improve the representation for PMLM.
Although it sheds light on the single sequence based methods for representation learning for proteins, more endeavors are needed to push forward the pre-training methods for protein sequences. Following the idea of PMLM, supervision from multiple residues, e.g. a Triple Masked Language Model, might also be helpful for pre-training the representation. The metaphor is that three points in Euclidean space follows the triangle in-equations inspired by AlphaFold2. However, the storage and computation cost will be cubic to the count of the masked tokens, thus how to efficiently pre-train the model might be an important issue to tackle, which will be left to future work.
6 Acknowledgments
We would like to thank Dahai Cheng for his valuable work on initial analysis.
References
- Alley et al. (2019) Ethan C Alley, Grigory Khimulya, Surojit Biswas, Mohammed AlQuraishi, and George M Church. Unified rational protein engineering with sequence-based deep representation learning. Nature Methods, 16(12):1315–1322, 2019.
- AlQuraishi (2019) Mohammed AlQuraishi. Alphafold at casp13. Bioinformatics, 35(22):4862–4865, 2019.
- Balakrishnan et al. (2011) Sivaraman Balakrishnan, Hetunandan Kamisetty, Jaime G Carbonell, Su-In Lee, and Christopher James Langmead. Learning generative models for protein fold families. Proteins: Structure, Function, and Bioinformatics, 79(4):1061–1078, 2011.
- Bepler & Berger (2019) Tristan Bepler and Bonnie Berger. Learning protein sequence embeddings using information from structure. In ICLR 2019 : 7th International Conference on Learning Representations, 2019.
- Bhattacharya et al. (2020) Nicholas Bhattacharya, Neil Thomas, Roshan Rao, Justas Daupras, Peter Koo, David Baker, Yun S Song, and Sergey Ovchinnikov. Single layers of attention suffice to predict protein contacts. bioRxiv, 2020.
- Boucher et al. (2016) Jeffrey I Boucher, Daniel NA Bolon, and Dan S Tawfik. Quantifying and understanding the fitness effects of protein mutations: Laboratory versus nature. Protein Science, 25(7):1219–1226, 2016.
- Consortium (2019) UniProt Consortium. Uniprot: a worldwide hub of protein knowledge. Nucleic acids research, 47(D1):D506–D515, 2019.
- Ekeberg et al. (2013) Magnus Ekeberg, Cecilia Lövkvist, Yueheng Lan, Martin Weigt, and Erik Aurell. Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models. Physical Review E, 87(1):012707, 2013.
- El-Gebali et al. (2019) Sara El-Gebali, Jaina Mistry, Alex Bateman, Sean R Eddy, Aurélien Luciani, Simon C Potter, Matloob Qureshi, Lorna J Richardson, Gustavo A Salazar, Alfredo Smart, et al. The pfam protein families database in 2019. Nucleic acids research, 47(D1):D427–D432, 2019.
- Elnaggar et al. (2020) A. Elnaggar, M. Heinzinger, C. Dallago, G. Rehawi, W. Yu, L. Jones, T. Gibbs, T. Feher, C. Angerer, M. Steinegger, D. Bhowmik, and B. Rost. ProtTrans: Towards cracking the language of lifes code through self-supervised deep learning and high performance computing. IEEE Transactions on Pattern Analysis and Machine Intelligence, (01):1–1, jul 2020. ISSN 1939-3539. doi: 10.1109/TPAMI.2021.3095381.
- Figliuzzi et al. (2016) Matteo Figliuzzi, Hervé Jacquier, Alexander Schug, Oliver Tenaillon, and Martin Weigt. Coevolutionary landscape inference and the context-dependence of mutations in beta-lactamase tem-1. Molecular biology and evolution, 33(1):268–280, 2016.
- Figliuzzi et al. (2018) Matteo Figliuzzi, Pierre Barrat-Charlaix, and Martin Weigt. How pairwise coevolutionary models capture the collective residue variability in proteins? Molecular biology and evolution, 35(4):1018–1027, 2018.
- Gloor et al. (2005) Gregory B Gloor, Louise C Martin, Lindi M Wahl, and Stanley D Dunn. Mutual information in protein multiple sequence alignments reveals two classes of coevolving positions. Biochemistry, 44(19):7156–7165, 2005.
- Hie et al. (2020) Brian Hie, Ellen Zhong, Bryan Bryson, and Bonnie Berger. Learning mutational semantics. In H. Larochelle, M. Ranzato, R. Hadsell, M. F. Balcan, and H. Lin (eds.), Advances in Neural Information Processing Systems, volume 33, pp. 9109–9121. Curran Associates, Inc., 2020. URL https://proceedings.neurips.cc/paper/2020/file/6754e06e46dfa419d5afe3c9781cecad-Paper.pdf.
- Hie et al. (2021a) Brian Hie, Ellen D. Zhong, Bonnie Berger, and Bryan Bryson. Learning the language of viral evolution and escape. Science, 371(6526):284–288, 2021a. ISSN 0036-8075. doi: 10.1126/science.abd7331. URL https://science.sciencemag.org/content/371/6526/284.
- Hie et al. (2021b) Brian L Hie, Kevin K Yang, and Peter S Kim. Evolutionary velocity with protein language models. bioRxiv, 2021b.
- Hopf et al. (2017) Thomas A Hopf, John B Ingraham, Frank J Poelwijk, Charlotta PI Schärfe, Michael Springer, Chris Sander, and Debora S Marks. Mutation effects predicted from sequence co-variation. Nature biotechnology, 35(2):128–135, 2017.
- Hsu et al. (2021) Chloe Hsu, Hunter Nisonoff, Clara Fannjiang, and Jennifer Listgarten. Combining evolutionary and assay-labelled data for protein fitness prediction. bioRxiv, 2021.
- Jones & Kandathil (2018) David T Jones and Shaun M Kandathil. High precision in protein contact prediction using fully convolutional neural networks and minimal sequence features. Bioinformatics, 34(19):3308–3315, 2018.
- Jones et al. (2012) David T Jones, Daniel WA Buchan, Domenico Cozzetto, and Massimiliano Pontil. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics, 28(2):184–190, 2012.
- Ju et al. (2021) Fusong Ju, Jianwei Zhu, Bin Shao, Lupeng Kong, Tie-Yan Liu, Wei-Mou Zheng, and Dongbo Bu. Copulanet: Learning residue co-evolution directly from multiple sequence alignment for protein structure prediction. Nature communications, 12(1):1–9, 2021.
- Jumper et al. (2021) John Jumper, Richard Evans, Alexander Pritzel, Tim Green, Michael Figurnov, Olaf Ronneberger, Kathryn Tunyasuvunakool, Russ Bates, Augustin Žídek, Anna Potapenko, et al. Highly accurate protein structure prediction with alphafold. Nature, pp. 1–11, 2021.
- Kamisetty et al. (2013) Hetunandan Kamisetty, Sergey Ovchinnikov, and David Baker. Assessing the utility of coevolution-based residue–residue contact predictions in a sequence-and structure-rich era. Proceedings of the National Academy of Sciences, 110(39):15674–15679, 2013.
- Kenton & Toutanova (2019) Jacob Devlin Ming-Wei Chang Kenton and Lee Kristina Toutanova. Bert: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of NAACL-HLT, pp. 4171–4186, 2019.
- Liu et al. (2019) Yinhan Liu, Myle Ott, Naman Goyal, Jingfei Du, Mandar Joshi, Danqi Chen, Omer Levy, Mike Lewis, Luke Zettlemoyer, and Veselin Stoyanov. Roberta: A robustly optimized bert pretraining approach. arXiv preprint arXiv:1907.11692, 2019.
- Lu et al. (2020) Amy X Lu, Haoran Zhang, Marzyeh Ghassemi, and Alan M Moses. Self-supervised contrastive learning of protein representations by mutual information maximization. BioRxiv, 2020.
- Madani et al. (2020) Ali Madani, Bryan McCann, Nikhil Naik, Nitish Shirish Keskar, Namrata Anand, Raphael R Eguchi, Po-Ssu Huang, and Richard Socher. ProGen: Language modeling for protein generation. arXiv preprint arXiv:2004.03497, 2020.
- Mann et al. (2014) Jaclyn K Mann, John P Barton, Andrew L Ferguson, Saleha Omarjee, Bruce D Walker, Arup Chakraborty, and Thumbi Ndung’u. The fitness landscape of hiv-1 gag: advanced modeling approaches and validation of model predictions by in vitro testing. PLoS computational biology, 10(8):e1003776, 2014.
- Martelotto et al. (2014) Luciano G Martelotto, Charlotte KY Ng, Maria R De Filippo, Yan Zhang, Salvatore Piscuoglio, Raymond S Lim, Ronglai Shen, Larry Norton, Jorge S Reis-Filho, and Britta Weigelt. Benchmarking mutation effect prediction algorithms using functionally validated cancer-related missense mutations. Genome biology, 15(10):1–20, 2014.
- Min et al. (2019) Seonwoo Min, Seunghyun Park, Siwon Kim, Hyun-Soo Choi, and Sungroh Yoon. Pre-training of deep bidirectional protein sequence representations with structural information. arXiv preprint arXiv:1912.05625, 2019.
- Morcos et al. (2011) Faruck Morcos, Andrea Pagnani, Bryan Lunt, Arianna Bertolino, Debora S Marks, Chris Sander, Riccardo Zecchina, José N Onuchic, Terence Hwa, and Martin Weigt. Direct-coupling analysis of residue coevolution captures native contacts across many protein families. Proceedings of the National Academy of Sciences, 108(49):E1293–E1301, 2011.
- Nambiar et al. (2020) Ananthan Nambiar, Maeve Heflin, Simon Liu, Sergei Maslov, Mark Hopkins, and Anna Ritz. Transforming the language of life: transformer neural networks for protein prediction tasks. In Proceedings of the 11th ACM International Conference on Bioinformatics, Computational Biology and Health Informatics, pp. 1–8, 2020.
- Perdigão et al. (2015) Nelson Perdigão, Julian Heinrich, Christian Stolte, Kenneth S. Sabir, Michael J. Buckley, Bruce Tabor, Beth Signal, Brian S. Gloss, Christopher J. Hammang, Burkhard Rost, Andrea Schafferhans, and Seán I. O’Donoghue. Unexpected features of the dark proteome. Proceedings of the National Academy of Sciences, 112(52):15898–15903, 2015. ISSN 0027-8424. doi: 10.1073/pnas.1508380112. URL https://www.pnas.org/content/112/52/15898.
- Peters et al. (2018) Matthew E Peters, Mark Neumann, Mohit Iyyer, Matt Gardner, Christopher Clark, Kenton Lee, and Luke Zettlemoyer. Deep contextualized word representations. In Proceedings of NAACL-HLT, pp. 2227–2237, 2018.
- Radford et al. (2018) Alec Radford, Karthik Narasimhan, Tim Salimans, and Ilya Sutskever. Improving language understanding by generative pre-training. 2018.
- Rao et al. (2019) Roshan Rao, Nicholas Bhattacharya, Neil Thomas, Yan Duan, Peter Chen, John Canny, Pieter Abbeel, and Yun Song. Evaluating Protein Transfer Learning with TAPE. In NeurIPS 2019 : Thirty-third Conference on Neural Information Processing Systems, pp. 9689–9701, 2019.
- Rao et al. (2020) Roshan Rao, Sergey Ovchinnikov, Joshua Meier, Alexander Rives, and Tom Sercu. Transformer protein language models are unsupervised structure learners. bioRxiv, 2020.
- Reva et al. (2011) Boris Reva, Yevgeniy Antipin, and Chris Sander. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Research, 39(17):e118–e118, 07 2011. ISSN 0305-1048. doi: 10.1093/nar/gkr407. URL https://doi.org/10.1093/nar/gkr407.
- Riesselman et al. (2017) Adam J Riesselman, John B Ingraham, and Debora S Marks. Deep generative models of genetic variation capture mutation effects. arXiv preprint arXiv:1712.06527, 2017.
- Rives et al. (2021) Alexander Rives, Joshua Meier, Tom Sercu, Siddharth Goyal, Zeming Lin, Jason Liu, Demi Guo, Myle Ott, C Lawrence Zitnick, Jerry Ma, et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proceedings of the National Academy of Sciences, 118(15), 2021.
- Seemayer et al. (2014) Stefan Seemayer, Markus Gruber, and Johannes Söding. CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics, 30(21):3128–3130, 2014.
- Senior et al. (2020) Andrew W Senior, Richard Evans, John Jumper, James Kirkpatrick, Laurent Sifre, Tim Green, Chongli Qin, Augustin Žídek, Alexander WR Nelson, Alex Bridgland, et al. Improved protein structure prediction using potentials from deep learning. Nature, 577(7792):706–710, 2020.
- Sheridan et al. (2015) Robert Sheridan, Robert J Fieldhouse, Sikander Hayat, Yichao Sun, Yevgeniy Antipin, Li Yang, Thomas Hopf, Debora S Marks, and Chris Sander. Evfold. org: Evolutionary couplings and protein 3d structure prediction. biorxiv, pp. 021022, 2015.
- Strodthoff et al. (2020) Nils Strodthoff, Patrick Wagner, Markus Wenzel, and Wojciech Samek. UDSMProt: universal deep sequence models for protein classification. Bioinformatics, 36(8):2401–2409, 2020.
- Sturmfels et al. (2020) Pascal Sturmfels, Jesse Vig, Ali Madani, and Nazneen Fatema Rajani. Profile prediction: An alignment-based pre-training task for protein sequence models. arXiv preprint arXiv:2012.00195, 2020.
- Vaswani et al. (2017) Ashish Vaswani, Noam Shazeer, Niki Parmar, Jakob Uszkoreit, Llion Jones, Aidan N Gomez, Lukasz Kaiser, and Illia Polosukhin. Attention is all you need. arXiv preprint arXiv:1706.03762, 2017.
- Vazquez et al. (2009) Esther Vazquez, Neus Ferrer-Miralles, Ramon Mangues, Jose L Corchero, Simo Schwartz Jr, Antonio Villaverde, et al. Modular protein engineering in emerging cancer therapies. Current pharmaceutical design, 15(8):893–916, 2009.
- Vig et al. (2020) Jesse Vig, Ali Madani, Lav R Varshney, Caiming Xiong, Richard Socher, and Nazneen Fatema Rajani. Bertology meets biology: Interpreting attention in protein language models. arXiv preprint arXiv:2006.15222, 2020.
- Wang et al. (2017) Sheng Wang, Siqi Sun, Zhen Li, Renyu Zhang, and Jinbo Xu. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS computational biology, 13(1):e1005324, 2017.
- Whitehead et al. (2012) Timothy A Whitehead, Aaron Chevalier, Yifan Song, Cyrille Dreyfus, Sarel J Fleishman, Cecilia De Mattos, Chris A Myers, Hetunandan Kamisetty, Patrick Blair, Ian A Wilson, et al. Optimization of affinity, specificity and function of designed influenza inhibitors using deep sequencing. Nature biotechnology, 30(6):543–548, 2012.
- Xiao et al. (2021) Yijia Xiao, Jiezhong Qiu, Ziang Li, Chang-Yu Hsieh, and Jie Tang. Modeling protein using large-scale pretrain language model. arXiv preprint arXiv:2108.07435, 2021.
- Xu (2019) Jinbo Xu. Distance-based protein folding powered by deep learning. Proceedings of the National Academy of Sciences, 116(34):16856–16865, 2019.
- Yang et al. (2020) Jianyi Yang, Ivan Anishchenko, Hahnbeom Park, Zhenling Peng, Sergey Ovchinnikov, and David Baker. Improved protein structure prediction using predicted interresidue orientations. Proceedings of the National Academy of Sciences, 117(3):1496–1503, 2020.