RIXS Reveals Hidden Local Transitions of the Aqueous OH Radical
Abstract
Resonant inelastic x-ray scattering (RIXS) provides remarkable opportunities to interrogate ultrafast dynamics in liquids. Here we use RIXS to study the fundamentally and practically important hydroxyl radical in liquid water, OH. Impulsive ionization of pure liquid water produced a short-lived population of OH, which was probed using femtosecond x-rays from an x-ray free-electron laser. We find that RIXS reveals localized electronic transitions that are masked in the ultraviolet absorption spectrum by strong charge-transfer transitions—thus providing a means to investigate the evolving electronic structure and reactivity of the hydroxyl radical in aqueous and heterogeneous environments. First-principles calculations provide interpretation of the main spectral features.
The hydroxyl radical (OH) is of major importance for atmospheric, astrochemical, biological, industrial, and environmental research. In the gas phase, OH is the primary oxidizing agent that rids the atmosphere of volatile organic compounds and other pollutants Isaksen and Dalsøren (2011). It is a key tracer describing the evolution and thermodynamics of interstellar clouds Rank et al. (1971). Despite its reactive nature stemming from an open electronic shell, the gas -phase absorption spectrum of OH has been fully characterized in the microwave Robinson and McGee (1967), infrared, optical/ultraviolet (UV) Huber and Herzberg (1979), and, more recently, the x-ray Stranges et al. (2002) spectral ranges. Beyond purely gas-phase processes, these OH fingerprints also characterize heterogeneous processes such as the generation of reactive oxygen species from photocatalysis Nosaka and Nosaka (2017).
Spectroscopic characterization of the hydroxyl radical in the condensed phase is more challenging, owing to its extreme reactivity and short lifetime. Of particular interest is the characterization of OH in aqueous environments, which impacts radiation biology Alizadeh and Sanche (2012) and chemistry Garrett et al. (2005). Given its unpaired spin, electron spin resonance techniques are a natural choice to detect the presence of OH, either directly or through spin traps, but only microsecond timescales are accessible Nosaka and Nosaka (2017). For faster timescales, desired for tracking reaction dynamics, one may consider UV spectroscopy. The UV spectrum of solvated OH obtained via pulsed radiolysis of water Czapski and Bielski (1993) is reproduced in Fig. 1. It is dominated by a strong feature at 230 nm (5.4 eV), whereas the dominant gas-phase absorption at 309 nm (4 eV), due to valence excitation from the ground (X) to the lowest excited electronic state (A), is barely visible. On the basis of electronic structure calculations Hamad et al. (2002); Chipman (2008, 2011); Codorniu-Hernandez et al. (2013), this dominant spectral feature of OH() was attributed to charge-transfer (CT) transitions from the lone pair of nearby waters, filling the hole in the OH orbital (Fig. 1, bottom left).
Resonant inelastic x-ray scattering (RIXS) delivers atomic-site specific information about the local electronic structure and dynamics in condensed phase. The application of soft x-ray RIXS to liquids Guo et al. (2002) has generated considerable attention; improvements in sensitivity Fuchs et al. (2008) and energy resolution Hennies et al. (2010) continue to open new perspectives on fundamental liquid-phase interactions Sun et al. (2011); Harada et al. (2013); Vaz da Cruz et al. (2019). Recently, the combination of RIXS, liquid microjets, and x-ray free-electron lasers enabled time-resolved measurements of electronic structure of transient species in solutions Wernet et al. (2015); Jay et al. (2018).
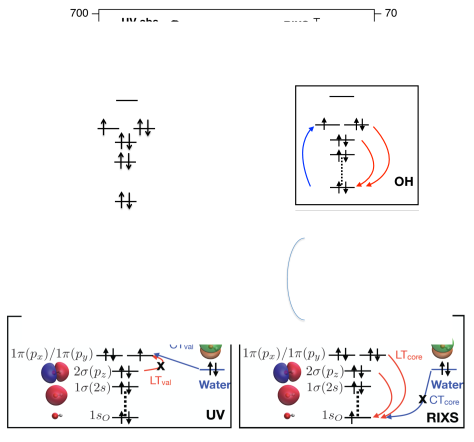
Here we report the RIXS spectrum for the short-lived OH radical in water (Fig. 1). In sharp contrast to the UV spectrum, the RIXS spectrum of OH features two peaks corresponding to transitions between the OH orbitals (Fig. 1, bottom right). The energy difference between the elastic and inelastic peaks corresponds to the XA transition, which, in turn, roughly equals the energy gap between the and orbitals. Thus, RIXS reveals intrinsic local electronic structure of solvated OH, which is obscured in the UV region by CT transitions. In contrast to CT transitions, which are characteristic of the solvent and its local structure, local transitions (LT) are fingerprints of the solute and can, therefore, be used to track reactive hydroxyl radicals in various complex and heterogeneous environments. The CT transitions in the RIXS spectrum are suppressed because the compact shape of the core orbital results in poor overlap with the lone pairs of neighboring waters. We confirm this by ab initio RIXS calculations using a new electronic structure method Vidal et al. (2019); Nanda et al. (2020) based on the equation-of-motion coupled-cluster (EOM-CC) theory. These calculations also reproduce the relative RIXS line intensities, positions, and widths for the elastic and inelastic peaks of OH and OH.
The OH spectra are also compared to the x-ray emission spectra (XES) of liquid water, where the role of hydrogen bonding and ultrafast dynamics has long been debated Fuchs et al. (2008); Tokushima et al. (2008); Harada et al. (2013); Pietzsch et al. (2015); Fransson et al. (2016); Yamazoe et al. (2019); Niskanen et al. (2019). If core-ionized water dissociates prior to core-hole decay, a core-excited OH is formed in the intermediate RIXS state (Fig.1). The lower-energy component of the water XES doublet has been attributed to this ultrafast dissociation Fuchs et al. (2008) and our measurement of the position of the OH RIXS resonance directly provides relevant information that previously was indirectly deduced Yamazoe et al. (2019).
We create the transient hydroxyl radical using strong-field ionization in pure liquid water Loh et al. (2020). The laser-induced ionization initially forms a water cation (H2O+), which undergoes ultrafast proton transfer with a neighboring water molecule on the sub-100-fs timescale Kamarchik et al. (2010) forming the hydroxyl radical and hydronium ion (H3O+). In the time window between proton transfer (100 fs) and geminate recombination, the ionized liquid water sample contains hydroxyl radicals. Because the transition in OH occurs cleanly in the “water window”, i.e., below the liquid water absorption edge, its kinetics can be readily probed via transient absorption Loh et al. (2020) and, simultaneously, via RIXS. Importantly, the latter allows investigation of local valence transitions, which are chemically most relevant.
Briefly, optical-pump x-ray-probe RIXS was performed using the Soft X-ray Research (SXR) instrument Schlotter et al. (2012) at the Linac Coherent Light Source (LCLS) at SLAC National Accelerator Laboratory. Monochromatized x-ray pulses were scanned from 518 to 542 eV (J, fs, 200 meV bandwidth). Three photon detection channels were simultaneously recorded: transmission, total fluorescence, and dispersed emission. A detailed description of the performance of the optical laser, water jet, and x-ray monochromator calibration, shot-by-shot normalization procedures can be found in Loh et al. (2020). Here we describe additionally the x-ray emission spectrometer and experimental geometries for RIXS measurements of OH.
X-ray emission was collected perpendicular to the incoming x-ray beam and along the x-ray polarization axis using a variable-line-spacing grating-based spectrometer Chuang et al. (2017). A CCD camera located at the exit plane of the spectrometer recorded images on a shot-by-shot basis.
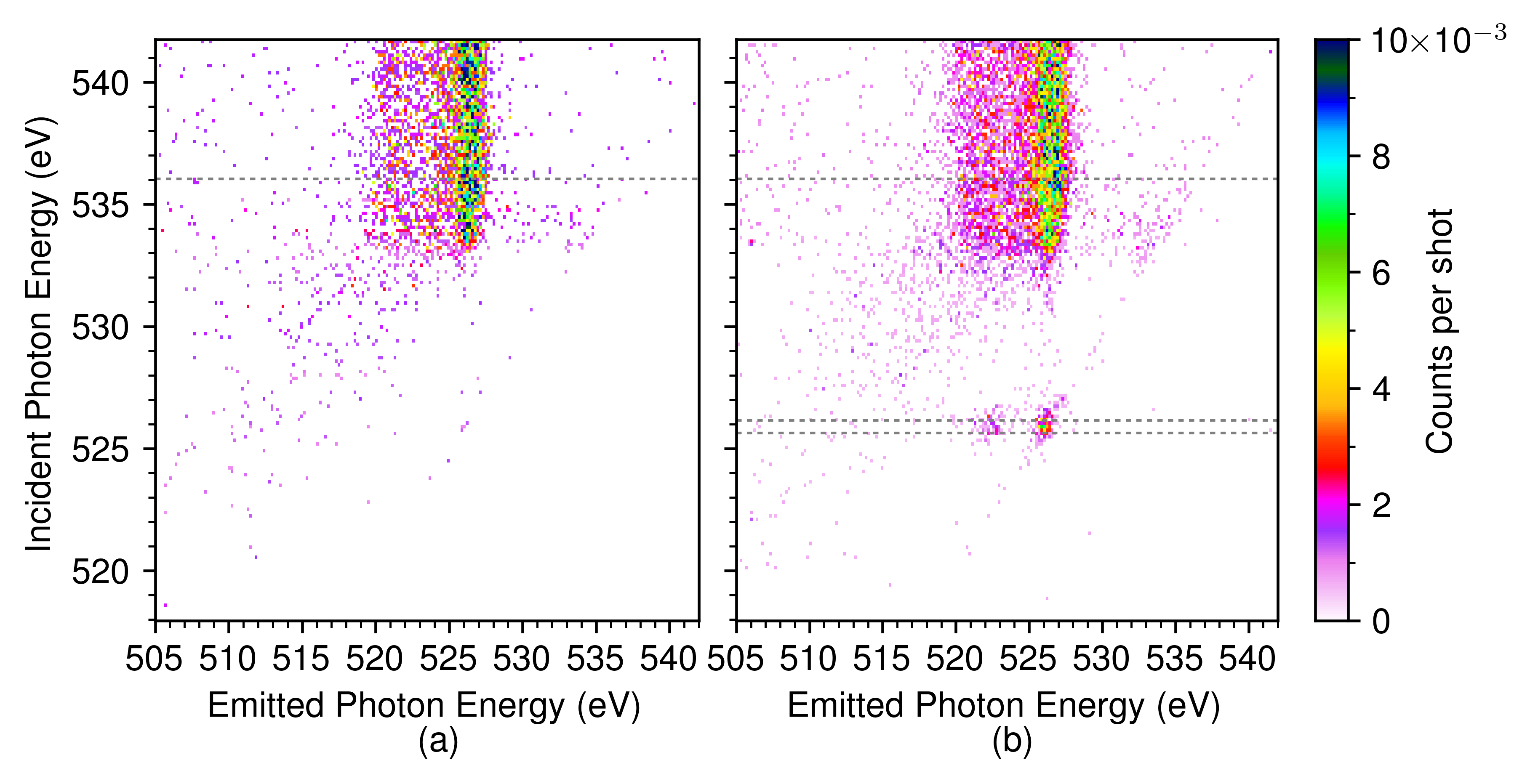
The energy dispersion and absolute energy of the incoming monochromatized radiation were previously calibrated Loh et al. (2020); Nagasaka et al. (2010). The dispersion of the emission spectrometer was determined by fitting a first degree polynomial to the elastic line visible in Fig. 2(b).
To gain insight into the nature of main spectral features, we carried out electronic structure calculations using EOM-CC Krylov (2008) with single and double excitations (EOM-CCSD), augmented by core-valence separation (CVS) Cederbaum et al. (1980) to enable access to core-level states Coriani and Koch (2015); Vidal et al. (2019). As a multistate method, EOM-CC treats different valence and core-level states on an equal footing and is particularly well suited for modeling molecular properties, including non-linear properties Krylov (2008); Helgaker et al. (2012); Nanda and Krylov (2015, 2018). To account for solvent effects, the spectral calculations of OH/OH were carried out within the QM/MM (quantum mechanics-molecular mechanics) scheme with water molecules described by classical force field and OH/OH- described by EOM-CCSD by using snapshots from equilibrium ab initio molecular dynamics simulations.
The electronic factors entering RIXS cross sections are the RIXS transition moments given by the following Kramers-Heisenberg-Dirac (KHD) expression Gel’mukhanov and Ågren (1999):
where and denote the initial and final electronic states (i.e., ground and valence excited state of OH), / are the incoming/outgoing photon frequencies, and the sum runs over all electronic states; is the energy difference between states and , and is the imaginary inverse lifetime parameter for state . In the present experiment, the dominant contribution to the RIXS cross section comes from the term corresponding to the state, which is resonant with excitation frequency of 526 eV, such that the spectra can be qualitatively understood within a three-states model. Within the EOM-CC framework, the KHD expression is evaluated using EOM-CC energies and wave functions. Rather than arbitrarily truncating the sum over states, we replace all s with a phenomenological damping factor and use damped response theory to convert the KHD expression into a numerically tractable closed form Nanda and Krylov (2015); Nanda et al. (2020); Faber and Coriani (2019). Robust convergence of the auxiliary response equations is achieved by using CVS within the damped response domain Nanda et al. (2020). The resulting method combines rigorous treatment of RIXS cross sections and high-level description of electron correlation. To describe vibrational structure in the RIXS spectrum, we computed Franck-Condon factors (FCFs) using three-states model (as was done in Ref. Sun et al. (2011)) and harmonic approximation. To quantify relative strengths of local and CT transitions, we also carried out calculations on model water-OH structures. All calculations were performed using the Q-Chem electronic structure package Krylov and Gill (2013). Full details of computational protocols are given in the SI.
Theoretical estimates of key structural parameters of the isolated OH given in Table 1 agree well with experimental values Huber and Herzberg (1979); Stranges et al. (2002). Theory overestimates the energy of the valence transition by 0.08 eV and underestimates the energy of the core-excited state by 0.7 eV; these differences are within the error bars of the method Vidal et al. (2019). The variations in bond lengths and frequencies among different states are consistent with the molecular orbital picture of the electronic states. The structural differences between the states give rise to a vibrational progression in the x-ray absorption spectrum; the computed FCFs are in excellent agreement with the experimental ones (see SI).
| State | Character | , eV | , Å | , eV | , a.u. | |
|---|---|---|---|---|---|---|
| X() | 0.000 | 0.972 | 0.468 | 0.701 | a | |
| 0.000 | 0.970 | 0.463 | b | |||
| core | 1s | 525.1 | 0.916 | 0.543 | 0.814 | a |
| 525.8 | 0.915 | 0.533 | c | |||
| A() | 4.128 | 1.014 | 0.398 | 0.801 | a | |
| 4.052 | 1.012 | 0.394 | b |
Fig. 2 shows the RIXS maps before and after the ionization pulse. The RIXS map prior to ionization, Fig. 2(a), is in agreement with earlier measurements Fuchs et al. (2008); Tokushima et al. (2008); Fransson et al. (2016). There is a threshold for emission at 534 eV excitation energy and a pre-edge peak at 535 eV. After ionization, Fig. 2(b), a new resonant feature appears at 526 eV excitation energy that is identified as transition of OH: its position is near that of gas-phase OH Stranges et al. (2002) and its kinetics are consistent with proton transfer Loh et al. (2020). The position of the quasi-elastic RIXS line of OH coincides with the lower-energy component of the water XES doublet (see dashed line in Fig. 3), providing a check on the absolute energy and consistent with the interpretation Fuchs et al. (2008) of this peak as due to ultrafast dissociation.
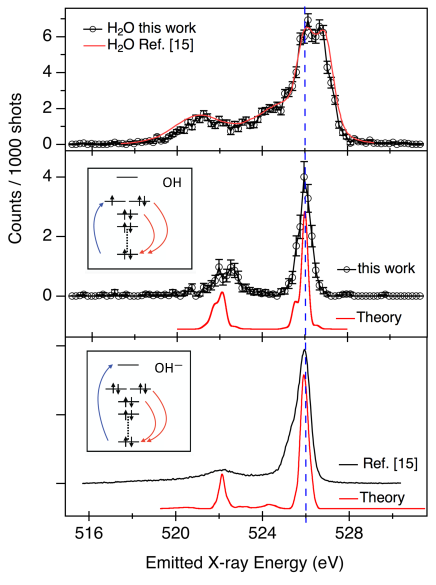
Table 2 summarises the main RIXS features of OH and OH, comparing experimental and theoretical values. The RIXS spectrum has a peak with 0.7 eV FWHM that assigned to quasi-elastic scattering to the electronic ground state. We also observe a 1.5 eV wide structure beginning at 3.6 eV energy loss that corresponds to scattering to the first electronically excited state: . The calculations reproduce the gap () between the quasi-elastic and energy-loss peak well; however, the absolute position of the transition is 1 eV off. EOM-EE-CCSD excitation spectra for core-level transitions often exhibit systematic shifts of 0.5-1.5 eV, attributed to insufficient treatment of electron correlation Vidal et al. (2019), while the relative positions of the peaks are reproduced with higher accuracy. The intensity ratio of the two peaks stems from their and character and is reproduced qualitatively by our calculations.
| Species | Source | , eV | , eV | Ratio |
|---|---|---|---|---|
| OH() | this worka | 526.0 | -3.8 | 2.1 |
| OH() | this workb | 525.0 | -4.0 | 2.04 |
| OH-() | Ref. 15. | 526.5 | -3.8 | 4.3 |
| OH-() | this workb | 526.2 | -3.8 | 5.20 |
a Experiment. b Theory.
Both quasi-elastic and energy-loss peaks are broadened due to the interaction with polar solvent and to vibrational structure. As shown in Table 1, the dipole moment in electronically excited OH is 14% larger than in the ground state, suggesting larger inhomogeneous broadening for the energy-loss peak; this is confirmed by our QM/MM calculations where the effect of the solvent is treated explicitly. The analysis of structural differences between the ground X(), valence excited A(), and core-excited states suggests longer vibrational progression for the energy-loss peak, which is confirmed by the computed FCFs (see SI). This trend can be rationalized by the shapes of molecular orbitals: the bonding character of orbital involved in the transition renders it more sensitive to vibrational excitation.
The middle and bottom panels of Fig. 3 compare the RIXS of OH to that of OH Fuchs et al. (2008). The two species show very similar emission spectra, as expected from the similarity of the intermediate RIXS state. There is a significant difference in the intensities of the quasi-elastic peak and the energy-loss () feature. The observed ratios for OH and OH are 2.1:1 and 4.3:1, compared to the calculated 2.04:1 and 5.20:1. In OH- calculations, we assumed resonant excitation to the lowest XAS peak of solvated OH-, which roughly corresponds to the transition to a diffuse -type orbital. The observed RIXS ratio from OH depends strongly on the nature of the intermediate state and, therefore, would be very sensitive to the excitation frequency; thus, the discrepancy between the computed values and Ref. Fuchs et al. (2008) could be due to different excitation regime.
To rationalize the apparent absence of the CTcore transitions in RIXS, we computed valence and core-level transitions for model OH-H2O structures. For the hemibonded structure, thought to be responsible for the CTval spectral feature in the UV-visible spectrum Chipman (2008, 2011), the oscillator strength for the local XA valence transition is 5 times smaller than that of the CTval transition. In contrast, the oscillator strength for the CTcore transition is 50 times smaller than that of the LTcore due to the poor overlap of the lone pair of water with the compact orbital of OH.
In summary, we have measured RIXS of the short-lived hydroxyl radical in pure liquid water. At the OH resonance of 526 eV, an energy-loss feature at 3.8 eV, corresponding to the localized XA transition of OH, was observed. The position of the OH resonance relative to bulk water XES provides information relevant to the long-standing debate on the structural versus dynamical interpretation of water XES. Ab initio calculations reproduce the positions, relative intensity, and broadening of the quasi-elastic and energy-loss peaks of OH() and OH-() and provide insight into the relative intensities of the local and CT RIXS transitions. Time-resolved RIXS, enabled by the availability of intense, tunable ultrafast x-ray pulses from XFELs, highlights the localized transition in this transient species, which is otherwise hidden in direct UV absorption spectra. This ability to report on intrinsic electronic structure of OH rather than on the properties of the solvent and its structure (which is revealed by the CT transitions dominating the UV spectrum) represents the key advantage of RIXS, demonstrating that it may be used to track ultrafast reactions of the chemically aggressive hydroxyl radical in aqueous and potentially more complex environments.
Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Science, Chemical Sciences, Geosciences and Biosciences Division that supported the Argonne group under contract number DE-AC02-06CH11357. Use of the Linac Coherent Light Source (LCLS), SLAC National Accelerator Laboratory, and, resources of the Center for Nanoscale Materials (CNM), Argonne National Laboratory, are supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences (BES) under Contracts DE-AC02-76SF00515 and DE-AC02-06CH11357. L.Y. acknowledges support from Laboratory Directed Research and Development (LDRD) funding from Argonne National Laboratory for conceptual design and proposal preparation. L.K., J.-E.R. acknowledge support from the Swedish Science Council (2018-04088). L.K. was also supported by the EuXFEL. Z.-H.L., T.D., and M.S.B.M.Y. acknowledge support from the Singapore Ministry of Education (MOE2014-T2-2-052, RG105/17 and RG109/18). M.S. was supported by the CNRS GotoXFEL program. C.A. and R.S. were supported by the Cluster of Excellence ‘Advanced Imaging of Matter’ of the Deutsche Forschungsgemeinschaft (DFG) - EXC 2056 - project ID 390715994. R.S. acknowledges support by the Chemical Sciences, Geosciences, and Biosciences Division, Office of Basic Energy Sciences, Office of Science, U.S. Department of Energy, Grant No. DE-SC0019451. M.L.V. and S.C. acknowledge support from DTU Chemistry (start-up PhD grant). S. C. acknowledges support from the Independent Research Fund Denmark – DFF-RP2 grant no. 7014-00258B. At USC, this work was supported by the U.S. National Science Foundation (No. CHE-1856342 to A.I.K.). A.I.K. is also a grateful recipient of the Simons Fellowship in Theoretical Physics and Mildred Dresselhaus Award from the Hamburg Centre for Ultrafast Imaging, which supported her sabbatical stay in Germany.
References
- Isaksen and Dalsøren (2011) I. S. A. Isaksen and S. B. Dalsøren, Science 331, 38 (2011).
- Rank et al. (1971) D. M. Rank, C. H. Townes, and W. J. Welch, Science 174, 1083 (1971).
- Robinson and McGee (1967) B. J. Robinson and R. X. McGee, Annual Review of Astronomy and Astrophysics 5, 183 (1967).
- Huber and Herzberg (1979) K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure— IV. Constants of Diatomic Molecules (Van Nostrand Reinhold, New York, 1979).
- Stranges et al. (2002) S. Stranges, R. Richter, and M. Alagia, The Journal of Chemical Physics 116, 3676 (2002).
- Nosaka and Nosaka (2017) Y. Nosaka and A. Y. Nosaka, Chemical Reviews 117, 11302 (2017).
- Alizadeh and Sanche (2012) E. Alizadeh and L. Sanche, Chemical Reviews 112, 5578 (2012).
- Garrett et al. (2005) B. C. Garrett, D. A. Dixon, D. M. Camaioni, D. M. Chipman, M. A. Johnson, C. D. Jonah, G. A. Kimmel, J. H. Miller, T. N. Rescigno, P. J. Rossky, S. S. Xantheas, S. D. Colson, A. H. Laufer, D. Ray, P. F. Barbara, D. M. Bartels, K. H. Becker, K. H. Bowen, S. E. Bradforth, I. Carmichael, J. V. Coe, L. R. Corrales, J. P. Cowin, M. Dupuis, K. B. Eisenthal, J. A. Franz, M. S. Gutowski, K. D. Jordan, B. D. Kay, J. A. LaVerne, S. V. Lymar, T. E. Madey, C. W. McCurdy, D. Meisel, S. Mukamel, A. R. Nilsson, T. M. Orlando, N. G. Petrik, S. M. Pimblott, J. R. Rustad, G. K. Schenter, S. J. Singer, A. Tokmakoff, L.-S. Wang, , and T. S. Zwier, Chemical Reviews 105, 355 (2005).
- Czapski and Bielski (1993) G. Czapski and B. H. Bielski, Radiation Physics and Chemistry 41, 503 (1993).
- Hamad et al. (2002) S. Hamad, S. Lago, and J. A. Mejías, The Journal of Physical Chemistry A 106, 9104 (2002).
- Chipman (2008) D. M. Chipman, The Journal of Physical Chemistry A 112, 13372 (2008).
- Chipman (2011) D. M. Chipman, The Journal of Physical Chemistry A 115, 1161 (2011).
- Codorniu-Hernandez et al. (2013) E. Codorniu-Hernandez, A. D. Boese, and P. G. Kusalik, Canadian Journal of Chemistry 91, 544 (2013).
- Guo et al. (2002) J.-H. Guo, Y. Luo, A. Augustsson, J.-E. Rubensson, C. Såthe, H. Ågren, H. Siegbahn, and J. Nordgren, Phys. Rev. Lett. 89, 137402 (2002).
- Fuchs et al. (2008) O. Fuchs, M. Zharnikov, L. Weinhardt, M. Blum, M. Weigand, Y. Zubavichus, M. Bär, F. Maier, J. D. Denlinger, C. Heske, M. Grunze, and E. Umbach, Phys. Rev. Lett. 100, 027801 (2008).
- Hennies et al. (2010) F. Hennies, A. Pietzsch, M. Berglund, A. Föhlisch, T. Schmitt, V. Strocov, H. O. Karlsson, J. Andersson, and J.-E. Rubensson, Phys. Rev. Lett. 104, 193002 (2010).
- Sun et al. (2011) Y.-P. Sun, F. Hennies, A. Pietzsch, B. Kennedy, T. Schmitt, V. N. Strocov, J. Andersson, M. Berglund, J.-E. Rubensson, K. Aidas, F. Gel’mukhanov, M. Odelius, and A. Föhlisch, Phys. Rev. B 84, 132202 (2011).
- Harada et al. (2013) Y. Harada, T. Tokushima, Y. Horikawa, O. Takahashi, H. Niwa, M. Kobayashi, M. Oshima, Y. Senba, H. Ohashi, K. T. Wikfeldt, A. Nilsson, L. G. M. Pettersson, and S. Shin, Phys. Rev. Lett. 111, 193001 (2013).
- Vaz da Cruz et al. (2019) V. Vaz da Cruz, F. Gel’mukhanov, S. Eckert, M. Iannuzzi, E. Ertan, A. Pietzsch, R. C. Couto, J. Niskanen, M. Fondell, M. Dantz, T. Schmitt, X. Lu, D. McNally, R. M. Jay, V. Kimberg, A. Föhlisch, and M. Odelius, Nature Communications 10, 1013 (2019).
- Wernet et al. (2015) P. Wernet, K. Kunnus, I. Josefsson, I. Rajkovic, W. Quevedo, M. Beye, S. Schreck, S. Grübel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, F. M. De Groot, K. J. Gaffney, S. Techert, M. Odelius, and A. Föhlisch, Nature 520, 78 (2015).
- Jay et al. (2018) R. M. Jay, J. Norell, S. Eckert, M. Hantschmann, M. Beye, B. Kennedy, W. Quevedo, W. F. Schlotter, G. L. Dakovski, M. P. Minitti, M. C. Hoffmann, A. Mitra, S. P. Moeller, D. Nordlund, W. Zhang, H. W. Liang, K. Kunnus, K. Kubiček, S. A. Techert, M. Lundberg, P. Wernet, K. Gaffney, M. Odelius, and A. Föhlisch, The Journal of Physical Chemistry Letters 9, 3538 (2018).
- Vidal et al. (2019) M. L. Vidal, X. Feng, E. Epifanovski, A. I. Krylov, and S. Coriani, J. Chem. Theory Comput. 15, 3117 (2019).
- Nanda et al. (2020) K. Nanda, M. L. Vidal, R. Faber, S. Coriani, and A. I. Krylov, Phys. Chem. Chem. Phys. 22, 2629 (2020).
- Tokushima et al. (2008) T. Tokushima, Y. Harada, O. Takahashi, Y. Senba, H. Ohashi, L. G. M. Pettersson, A. Nilsson, and S. Shin, Chemical Physics Letters 460, 387 (2008).
- Pietzsch et al. (2015) A. Pietzsch, F. Hennies, P. S. Miedema, B. Kennedy, J. Schlappa, T. Schmitt, V. N. Strocov, and A. Föhlisch, Phys. Rev. Lett. 114, 088302 (2015).
- Fransson et al. (2016) T. Fransson, Y. Harada, N. Kosugi, N. A. Besley, B. Winter, J. J. Rehr, L. G. M. Pettersson, and A. Nilsson, Chemical Reviews 116, 7551 (2016).
- Yamazoe et al. (2019) K. Yamazoe, J. Miyawaki, H. Niwa, A. Nilsson, and Y. Harada, The Journal of Chemical Physics 150, 204201 (2019).
- Niskanen et al. (2019) J. Niskanen, M. Fondell, C. J. Sahle, S. Eckert, R. M. Jay, K. Gilmore, A. Pietzsch, M. Dantz, X. Lu, D. E. McNally, T. Schmitt, V. Vaz da Cruz, V. Kimberg, F. Gel’mukhanov, and A. Föhlisch, Proceedings of the National Academy of Sciences 116, 4058 (2019).
- Loh et al. (2020) Z.-H. Loh, G. Doumy, C. Arnold, L. Kjellsson, S. H. Southworth, A. Al Haddad, Y. Kumagai, M.-F. Tu, P. J. Ho, A. M. March, R. D. Schaller, M. S. Bin Mohd Yusof, T. Debnath, M. Simon, R. Welsch, L. Inhester, K. Khalili, K. Nanda, A. I. Krylov, S. Moeller, G. Coslovich, J. Koralek, M. P. Minitti, W. F. Schlotter, J.-E. Rubensson, R. Santra, and L. Young, Science 367, 179 (2020).
- Kamarchik et al. (2010) E. Kamarchik, O. Kostko, J. M. Bowman, M. Ahmed, and A. I. Krylov, J. Chem. Phys. 132, 194311 (2010).
- Schlotter et al. (2012) W. F. Schlotter, J. J. Turner, M. Rowen, P. Heimann, M. Holmes, O. Krupin, M. Messerschmidt, S. Moeller, J. Krzywinski, R. Soufli, M. Fernández-Perea, N. Kelez, S. Lee, R. Coffee, G. Hays, M. Beye, N. Gerken, F. Sorgenfrei, S. Hau-Riege, L. Juha, J. Chalupsky, V. Hajkova, A. P. Mancuso, A. Singer, O. Yefanov, I. A. Vartanyants, G. Cadenazzi, B. Abbey, K. A. Nugent, H. Sinn, J. Lüning, S. Schaffert, S. Eisebitt, W.-S. Lee, A. Scherz, A. R. Nilsson, and W. Wurth, Review of Scientific Instruments 83, 043107 (2012).
- Chuang et al. (2017) Y.-D. Chuang, Y.-C. Shao, A. Cruz, K. Hanzel, A. Brown, A. Frano, R. Qiao, B. Smith, E. Domning, S.-W. Huang, L. A. Wray, W.-S. Lee, Z.-X. Shen, T. P. Devereaux, J.-W. Chiou, W.-F. Pong, V. V. Yashchuk, E. Gullikson, R. Reininger, W. Yang, J. Guo, R. Duarte, and Z. Hussain, Review of Scientific Instruments 88, 013110 (2017).
- Nagasaka et al. (2010) M. Nagasaka, T. Hatsui, T. Horigome, Y. Hamamura, and N. Kosugi, Journal of Electron Spectroscopy and Related Phenomena 177, 130 (2010).
- Krylov (2008) A. I. Krylov, Annu. Rev. Phys. Chem. 59, 433 (2008).
- Cederbaum et al. (1980) L. S. Cederbaum, W. Domcke, and J. Schirmer, Phys. Rev. A 22, 206 (1980).
- Coriani and Koch (2015) S. Coriani and H. Koch, J. Chem. Phys. 143, 181103 (2015).
- Helgaker et al. (2012) T. Helgaker, S. Coriani, P. Jørgensen, K. Kristensen, J. Olsen, and K. Ruud, Chem. Rev. 112, 543 (2012).
- Nanda and Krylov (2015) K. D. Nanda and A. I. Krylov, J. Chem. Phys. 142, 064118 (2015).
- Nanda and Krylov (2018) K. Nanda and A. I. Krylov, J. Chem. Phys. 149, 164109 (2018).
- Gel’mukhanov and Ågren (1999) F. Gel’mukhanov and H. Ågren, Phys. Rep. 312, 87 (1999).
- Faber and Coriani (2019) R. Faber and S. Coriani, Journal of Chemical Theory and Computation 15, 520 (2019).
- Krylov and Gill (2013) A. I. Krylov and P. M. W. Gill, WIREs: Comput. Mol. Sci. 3, 317 (2013).