ShortCake: An integrated platform for efficient and reproducible single-cell analysis
Abstract
Motivation: Recent advances in single-cell analysis have introduced new computational challenges. Researchers often need to use multiple analysis tools written in different programming languages while managing version conflicts between related packages within a single workflow. For the research community, minimizing the time spent on environment setup and installation issues is essential.
Results: We present ShortCake, a containerized platform that integrates a suite of single-cell analysis tools written in R and Python. ShortCake isolates competing Python tools into separate virtual environments that can be easily accessed within a Jupyter notebook. This enables users to effortlessly transition between various environments, including R, even within a single notebook.
Additionally, ShortCake offers multiple “flavors,” enabling users to select container images tailored to their specific needs. ShortCake provides a unified environment with fixed versions of various tools, thus streamlining workflows, reducing setup time, and improving reproducibility.
Availability and implementation: The ShortCake image is available on DockerHub
(https://hub.docker.com/r/rnakato/shortcake). The source code is available on GitHub (https://github.com/rnakato/ShortCake).
1 Introduction
Single-cell analysis is a powerful method for exploring cellular heterogeneity, lineage, and spatial information by profiling the transcriptome and epigenome of individual cells (Lareau et al. (2019); Stuart et al. (2019b)). The landscape of single-cell analysis is rapidly expanding. Currently, even for single-cell RNA sequencing (scRNA-seq) alone, more than 1,800 tools are registered in the scRNA-tools database (Zappia et al. (2018)). As a result, it has become common to combine multiple tools within a single project, or to test and compare different methods for the same analytical step, while working in multiple programming languages. Managing these heterogeneous environments can pose significant challenges, especially for researchers without a bioinformatics background.
Several pipelines have been developed to facilitate such integrated single-cell data analysis using multiple tools. For instance, the Seurat ecosystem in R and the scverse in Python offer valuable frameworks for single-cell analysis (Hao et al. (2024); Virshup et al. (2023)). However, these ecosystems do not provide environments for both R and Python, and do not solve installation problems. Users often struggle with dependency conflicts, version mismatches, and complex setup procedures when trying to combine various tools for their own custom analysis workflows. Although package managers like Conda (https://anaconda.org/) are helpful, they still have difficulty reconciling conflicting package dependencies. These problems can prevent researchers from carrying out their research ideas.
Another challenge is reproducibility, especially across different computational environments. For instance, identical workflows have produced different sets of detected transcripts on macOS versus Linux (Di Tommaso et al. (2017)). Improving the reproducibility of previous studies has been a key challenge.
To address these challenges, we developed ShortCake, a Docker-based platform that consolidates various single-cell RNA-seq and ATAC-seq (scATAC-seq) analysis tools in both R and Python environments, as well as correlated command-line tools. ShortCake separates conflicting Python packages into separate virtual environments, each of which is accessible via a Jupyter Notebook kernel. This streamlines the process of switching between tools without exiting an interactive session. ShortCake also provides multiple “flavors” of the image, allowing users to download only the necessary components and conserve computational resources. Additionally, since the ShortCake Dockerfile is publicly available, users can also edit it to build custom images that include any necessary additional tools. This architecture dramatically reduces installation costs and facilitates reproducibility among users and host computers. It lowers the barrier to entry for researchers and promotes reproducibility through standardized environments.
2 Design and Implementation
2.1 Overall architecture
Shortcake is distributed as a Docker image built on an Ubuntu 22.04 base layer. Docker packages software and its dependencies into lightweight containers, ensuring that the same environment runs identically on any host computer. To enable GPU computation, the latest version of Shortcake (v3.3.0) uses the CUDA 11.8.0 runtime and cuDNN 8 for Ubuntu 22.04. R is installed directly in the container image (v3.3.0 ships with R 4.4.1). Python environments are managed with Micromamba (https://github.com/mamba-org/micromamba-releases), and each package in the base environment is version-pinned via an env.yaml file.
ShortCake (v3.3.0) includes over 90 tools for single-cell analysis. These tools cover various steps, including quality control, doublet detection, batch integration, trajectory/velocity inference, spatial analysis, multimodal integration, and network reconstruction. ShortCake also includes reference genomes, gene annotations, and related demo datasets. See Supplementary Table 1 for a complete list.
2.2 Jupyter Notebook execusion
The recommended workflow is to start ShortCake’s Docker container, launch Jupyter Notebook, and connect through a web browser, for example:
docker run --rm -p 8888:8888 rnakato/shortcake jupyternotebook.sh
Users can also run the container on a remote server and access it from their local laptop.
ShortCake resolves conflicts among Python-based tools by assigning each tool to its own virtual environment. Tools that invoke other tools internally are bundled into the same environment. For instance, UnitVelo (Gao et al. (2022)), which depends on scvi-tools (Gayoso et al. (2022)), shares the same virtual environment.
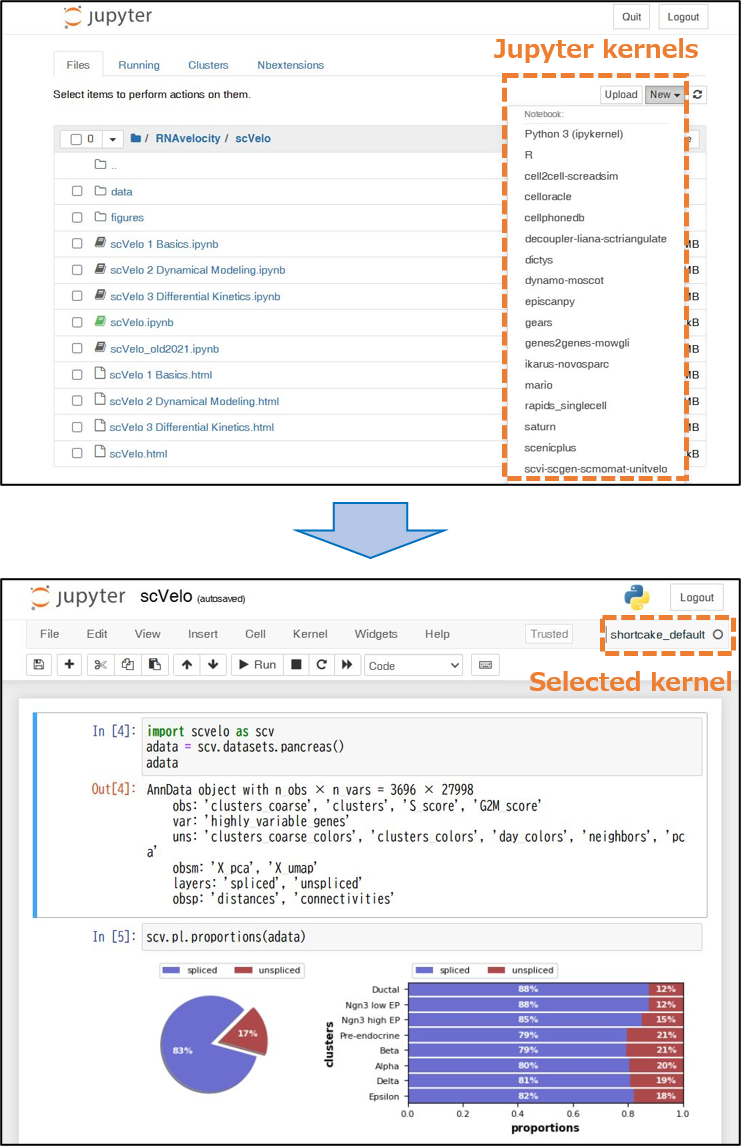
To streamline access to the many environments, ShortCake registers a dedicated Jupyter kernel for each, making them selectable within the Jupyter Notebook (Figure 1). This design enables users to effortlessly switch between environments within a single notebook and facilitates workflows that rely on multiple tools with conflicting dependencies. By clicking the “New” button in Jupyter Notebook and selecting the desired kernel, users can launch an analysis session that runs inside their chosen virtual environment.
2.3 Rstudio execusion
Although the R environment can also be invoked in Jupyter Notebook, certain R-based tools do not run properly in the interface (e.g., the interactive functions of Monocle3 (Qiu et al. (2017))). Shortcake can launch RStudio (https://docs.posit.co/ide/user/) with this command:
docker run --rm -p 8888:8888 rnakato/shortcake rstudio
so that users can begin their analyses in the RStudio environment, as all of the R libraries in Shortcake are already installed.
2.4 Command-line excusion
Several single-cell tools provide command-line tools. For example, Velocyto (Stuart et al. (2019a)) provides the command velocyto run10x to generate a .loom file. It can be executed as follows:
docker run --rm -p 8888:8888 rnakato/shortcake \ velocyto run10x -m repeat_msk.gtf mypath/genes.gtf
It is also possible to log directly into the ShortCake container and work inside it using the command-line interface.
docker run --rm -p 8888:8888 rnakato/shortcake /bin/bash
2.5 ShortCake flavors
One drawback of ShortCake is that the Docker image grows rapidly when it contains many virtual environments. Deep-learning tools that require CUDA libraries tend to be particularly large, often adding more than 10 GB to a single virtual environment. Consequently, the ShortCake image with the full model exceeds 100 GB on Docker, which is impractical for most laptops.
To circumvent this problem, we have prepared several “flavors” of ShortCake. By providing lightweight images that only include the set of tools most users need, it is easier to use for purposes such as tutorials. The main flavors are outlined below:
-
•
shortcake_seurat: Contains only Seurat (Hao et al. (2024)) and its related packages.
-
•
shortcake_r: Builds on shortcake_seurat with additional R packages. Jupyter Notebook is available, but no Python single-cell tools are installed.
- •
-
•
shortcake: Extends shortcake_light with nearly all remaining Python virtual environments.
-
•
shortcake_full: The comprehensive image, including every supported tool.
2.6 Customization and extensibility
Users can extend ShortCake either by modifying its original Dockerfile or by creating a new Dockerfile that begins with ’FROM rnakato/shortcake’. This flexibility enables users to incorporate the latest tools and their own scripts, making it easy to create custom analysis pipelines with ShortCake.
3 Discussion
ShortCake has been continuously updated since its inception in 2022 and has been utilized in several studies (Nagai et al. (2023); Shibata et al. (2024)). Its Docker-based approach enables users to replicate an identical analysis environment on any local machine with minimal effort, ensuring reproducible workflows. When Docker privileges are unavailable (e.g., on shared cluster servers), the same image can be run with Singularity (Kurtzer et al. (2017)) instead.
Community initiatives, such as nf-core (Marques de Almeida et al. ), have successfully standardized single-cell workflows. These initiatives provide curated Nextflow pipelines that can be executed reproducibly. This type of framework is ideal for large consortia that must run a single, agreed-upon pipeline on numerous samples in a tightly controlled environment. Conversely, ShortCake is designed for individual researchers or small groups exploring suitable case-specific workflows by iterating over alternative tools, parameter settings, and custom scripts. Rather than enforcing a fixed workflow, ShortCake offers a flexible interface that accelerates the prototyping phase for specific biological questions.
As the single-cell research field continues to advance, new technologies will emerge, such as spatial and perturbation analyses. We aim to keep up with these advances by providing the research community with well-validated tools that will facilitate their work.
4 Competing interests
No competing interest is declared.
5 Author contributions
R.N. developed ShortCake. R.N. and L.A.E.N. maintained and tested it. R.N. and L.A.E.N. wrote and revised the manuscript.
6 Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research under grant number 23H02466, the Japan Agency for Medical Research and Development under grant number JP23gm6310012h0004, and the JST FOREST Program under grant number JPMJFR224Y.
References
- Bergen et al. [2020] Volker Bergen, Marius Lange, Stefan Peidli, F. Alexander Wolf, and Fabian J. Theis. Generalizing RNA velocity to transient cell states through dynamical modeling. Nature Biotechnology, 38(12):1408–1414, 2020. doi:10.1038/s41587-020-0591-3.
- Di Tommaso et al. [2017] Paolo Di Tommaso, Maria Chatzou, Evan W. Floden, Pablo Prieto Barja, Emilio Palumbo, and Cedric Notredame. Nextflow enables reproducible computational workflows. Nature Biotechnology, 35(4):316–319, April 2017. doi:10.1038/nbt.3820.
- Gao et al. [2022] Mingze Gao, Chen Qiao, and Yuanhua Huang. Unitvelo: temporally unified RNA velocity reinforces single-cell trajectory inference. Nature Communications, 13:6586, 2022. doi:10.1038/s41467-022-34188-7.
- Gayoso et al. [2022] Adam Gayoso, Romain Lopez, Galen Xing, Pierre Boyeau, Valeh Valiollah Pour Amiri, Justin Hong, Katherine Wu, Michael Jayasuriya, Edouard Mehlman, Maxime Langevin, and et al. A Python library for probabilistic analysis of single-cell omics data. Nature Biotechnology, 40(2):163–166, 2022. doi:10.1038/s41587-021-01206-w.
- Hao et al. [2024] Yuhan Hao, Tim Stuart, Madeline H. Kowalski, Saket Choudhary, Paul Hoffman, Austin Hartman, Avi Srivastava, Gesmira Molla, Shaista Madad, Carlos Fernandez-Granda, and Rahul Satija. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nature Biotechnology, 42(2):293–304, 2024. doi:10.1038/s41587-023-01767-y.
- Kurtzer et al. [2017] Gregory M. Kurtzer, Vanessa Sochat, and Michael W. Bauer. Singularity: Scientific containers for mobility of compute. PLOS ONE, 12(5):e0177459, 2017. doi:10.1371/journal.pone.0177459.
- Lareau et al. [2019] Caleb A. Lareau, Felix M. Duarte, Graham Chew, Vinay K. Kartha, Zachary D. Burkett, et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nature Biotechnology, 37(8):916–924, 2019. doi:10.1038/s41587-019-0147-6.
- [8] Felipe Marques de Almeida, Alexander Peltzer, Gregor Sturm, Olga Botvinnik, Dongze He, Nico Trummer, Kevin Menden, Adam Talbot, Robert Syme, Maxime U. Garcia, Harshil Patel, Tom Kelly, Peter Bailey, Sangram K. Sahu, and nf-core community. nf-core/scrnaseq: 4.0.0. URL https://doi.org/10.5281/zenodo.15004569. Software.
- Nagai et al. [2023] Hiroki Nagai, Luis A. E. Nagai, Sohei Tasaki, Ryuichiro Nakato, Daiki Umetsu, Erina Kuranaga, Masayuki Miura, and Yuichiro Nakajima. Nutrient-driven dedifferentiation of enteroendocrine cells promotes adaptive intestinal growth in Drosophila. Developmental Cell, 58(18):1764–1781.e10, 2023. doi:10.1016/j.devcel.2023.08.022.
- Qiu et al. [2017] Xiaojie Qiu, Qi Mao, Ying Tang, Li Wang, Raghav Chawla, Hannah A. Pliner, and Cole Trapnell. Reversed graph embedding resolves complex single-cell trajectories. Nature Methods, 14(10):979–982, 2017. doi:10.1038/nmeth.4402.
- Shibata et al. [2024] Shun Shibata, Shun Endo, Luis A. E. Nagai, Eri H. Kobayashi, Atsushi Oike, Noriyuki Kobayashi, Aki Kitamura, Takahiro Hori, Yasuhito Nashimoto, Ryuichiro Nakato, Hirokazu Hamada, Hideki Kaji, Chisato Kikutake, Motoharu Suyama, Michiko Saito, Naoki Yaegashi, Hiroki Okae, and Tomoko Arima. Modeling embryo–endometrial interface recapitulating human embryo implantation. Science Advances, 10(8):eadi4819, 2024. doi:10.1126/sciadv.adi4819.
- Stuart et al. [2019a] Tim Stuart, Andrew Butler, Paul Hoffman, Christoph Hafemeister, Efthymia Papalexi, William M. Mauck, Yuhan Hao, Marlon Stoeckius, Peter Smibert, and Rahul Satija. Comprehensive integration of single-cell data. Nature, 177:1888–1902.e21, 2019a. doi:10.1038/s41586-018-0414-6.
- Stuart et al. [2019b] Tim Stuart, Andrew Butler, Paul Hoffman, Christoph Hafemeister, Efthymia Papalexi, William M. III Mauck, et al. Comprehensive integration of single-cell data. Cell, 177(7):1888–1902.e21, 2019b. doi:10.1016/j.cell.2019.05.031.
- Virshup et al. [2023] Isaac Virshup, Danila Bredikhin, Lukas Heumos, Giovanni Palla, Gregor Sturm, Adam Gayoso, Ilia Kats, Mikaela Koutrouli, Bonnie Berger, Dana Pe’er, Aviv Regev, Sarah A. Teichmann, Francesca Finotello, F. Alexander Wolf, Nir Yosef, Oliver Stegle, Fabian J. Theis, and The scverse Community. The scverse project provides a computational ecosystem for single-cell omics data analysis. Nature Biotechnology, 41(5):604–606, 2023. doi:10.1038/s41587-023-01733-8.
- Wolf et al. [2018] Florian A. Wolf, Philipp Angerer, and Fabian J. Theis. Scanpy: large-scale single-cell gene expression data analysis. Genome Biology, 19(1):15, 2018. doi:10.1186/s13059-017-1382-0.
- Zappia et al. [2018] Luke Zappia, Belinda Phipson, and Alicia Oshlack. Exploring the single-cell RNA-seq analysis landscape with the scrna-tools database. PLOS Computational Biology, 14(6):e1006245, 2018. doi:10.1371/journal.pcbi.1006245.